سلام همه! دخترانی که در چنین شرایطی بودند، پاسخ می دهند! در 27 مه، اولین نمایشگاه برگزار شد. سونوگرافی همه طبیعی بود. آنها این گوشی را فقط در مورد ثبت نام کردند، اما انتظار نداشتند که آنها بتوانند تماس بگیرند، و در حال حاضر یک هفته یک تماس - در CPS به عقب برگردد، خطر بالایی دارید. من خودم را به یاد نمی آورم، در اشک ها، من در پاهای پنبه ای گرفتم، تمام قطعات کاغذ را گرفتم. خطر 1:53 روز بعد من رفتم دو برابر شدم سونوگرافی به مدت زمان بسیار طولانی به معده نگاه کرد و به طور واضح، او چندین بار داپلر را شامل می شد، و همه چیز به نظر می رسد هر چیزی است، اما داپلرمتری شیر Truskupidal را دوست ندارد: Regurgitation. داده های سونوگرافی جدید در برنامه وارد شده و نتایج غربالگری محدودیت های هفتگی را وارد کرد، کامپیوتر یک خطر SD 1: 6 را صادر کرد. فرستاده شده به ژنتیک نگاهی به نتیجه گیری، او به من توضیح داد که این رگبار می تواند به سادگی یک ویژگی از جنین باشد، اما به اتهام با یک شاخص کاهش یافته PAPP-A - 0.232 مادر، نشانگر ناهنجاری های کروموزومی است. هر چیز دیگری در محدوده طبیعی است. آنها پیشنهاد کردند که بیوپسی کوروین بیوپسی را تصویب کنند. من هنوز رد شدم، پرستار تقریبا از صندلی سقوط کرد، مانند ریسک بسیار قد بلند است و هکتار درمان نمی شود و در جای من او حتی فکر نمی کند از یک دقیقه. علاقه مند به ژنتیک در مورد تجزیه و تحلیل پانورامای پانوراما (تجزیه و تحلیل ژنتیک عزیزم. تجزیه و تحلیل خون مادر)، او به من پاسخ داد که مطمئنا می تواند انجام شود، اما تنها 5 هکتار اصلی و چندین نادر را از بین می برد، او نمیتواند به طور کامل یک ناهنجاری را حذف کند و در مورد من تهاجم توصیه شده است. من قبلا یک تن از مقالات، سوالات و همه چیز را مانند این موضوع در این موضوع خوانده ام، و من نمی فهمم که در تجزیه و تحلیل های من خیلی وحشتناک است؟ Regurgitation به عنوان معلوم شد که در این دوره فیزیولوژیک است و به 18-20 هفته صورت می گیرد (اگر آن را در مورد خطر نقص های قلب صحبت نمی کند، بسیاری از آنها پس از زایمان عبور می کند، و برخی افراد با آن زندگی می کنند و هیچ چیز را تحت تاثیر قرار نمی دهد. به ویژه در شوهرش پروپس شیر مینرال که از مادر گرفته شده بود، شاید به نحوی متصل شود). هورمون ها ممکن است در همه چیز نشان داده نشوند، زیرا من از ابتدای بارداری توسط Duphaston پذیرفته شده ام، من 2 ساعت قبل از تجزیه و تحلیل (به نظر می رسد که شما نمی توانید به مدت 4 ساعت قبل از غذا بخورید، من در مورد آن نگفتم)، من قهوه، عصبی و نگرانی در مورد اولتراسوند و خون می ترسد، و اخیرا خستگی مزمن، با فرزند بزرگتر خسته می شود. و این همه بر نتایج تاثیر می گذارد. هیچ چیز مانند ژنتیک خواسته نشد، من علاقه مند نبودم، آنها نوعی نوار نقاله وجود داشت، و به نظر می رسید که برای آمار وجود دارد. اما تنها شک من آنها را در من کاشت، من فرار کردم، من در مورد یک سال پیش از آن نگران بودم. شوهر بیوپسی را متقاعد می کند. من وحشتناکی از عواقب می ترسم، من می ترسم از دست دادن یا آسیب رساندن به کودک، به خصوص اگر او سالم باشد. از یک طرف، اگر همه چیز خوب باشد، با تسکین دادن و ارسال همه پزشکان دور. از سوی دیگر، اگر همه چیز بد باشد، چه کاری باید انجام دهید؟ آیا می توانم بارداری را متوقف کنم، کودک شما را درون من قرار دهم، به ویژه در حال حاضر زمانی که به نظر می رسد، من شروع به احساس آن می کنم. اما یکی دیگر از گزینه ها قادر به افزایش چنین کودکانی است که نیاز به یک رویکرد خاص و توجه زیادی خواهد داشت، زمانی که گاهی اوقات می خواهد از یک دختر سالم فرار کند ... لعنتی، همه این افکار به من فرستاده می شوند. من نمی دانم چگونه می توانم ... فقط در صورتی که من داده های غربالگری را ارائه دهم:
مدت B-: 13
ضربان قلب 161 یخ / دقیقه
کانال های وریدی PI 1،160
Chorion / Planter پایین روی دیوار جلو
توتوینا 3 کشتی
آناتومی جنین: همه چیز تعیین می شود، همه چیز طبیعی است
b-HGCH 1،091 مامان
PAPP-A 0.232 مادر
رحم هنری PI 1،240 مامان
Trisomy 21 1: 6
Trisomy 18 1: 311
Trisomy 13 1: 205
پره اکلامپسی تا 34 هفته 1: 529
پره اکلامپسی تا 37 هفته 1: 524
7.1 طبقه بندی دیابت
دیابت(SD) یک گروه از بیماری های متابولیک مشخص شده توسط هیپرگلیسمی به علت نقض ترشح و / یا اثربخشی انسولین است. هیپرگلیسمی مزمن، در حال توسعه در SD، همراه با توسعه عوارض توسط بسیاری از ارگان ها و سیستم ها، اول از همه، از قلب، عروق خونی، چشم، کلیه ها و اعصاب همراه است. در مجموع 5-6 درصد از جمعیت رنج می برند. در کشورهای توسعه اقتصادی از لحاظ اقتصادی جهان هر 10-15 سال، تعداد بیماران مبتلا به CD به 2 بار افزایش می یابد. امید به زندگی در CD 10-15٪ کاهش می یابد.
علل توسعه SD به طور گسترده ای متفاوت است. در اکثریت قریب به اتفاق موارد CD، یا به علت کمبود انسولین مطلق (دیابت نوع 1 -SD-1) یا به علت کاهش حساسیت بافت های محیطی به انسولین در ترکیب با اختلال عملکرد اسرار سلول های بتا پانکراس (دیابت نوع 2 -SD-2). در برخی موارد، تخصیص بیمار به SD-1 یا SD-2 دشوار است، با این وجود، جبران CD بیشتر قابل توجه است، و نه دقیق بودن نوع آن. طبقه بندی اتئولوژیک چهار کلاس اصلی بالینی SD را اختصاص می دهد (جدول 7.1).
شایع ترین SD-1 (بند 7.5)، SD-2 (ص. 7.6) و SD بارداری (بند 7.9) در فصل های جداگانه مورد بحث قرار گرفته است. در انواع خاص دیگرتنها حدود 1٪ موارد SD وجود دارد. علت و پاتوژنز این نوع SD به نظر می رسد در مقایسه با SD-1 و به ویژه SD-2 مورد مطالعه قرار گیرد. تعدادی از انواع SD به علت به ارث برده اند نقایص ژنتیکی عملکردβ بطریاین شامل گزینه های مختلف برای سندرم Mody Mody به ارث برده شده در اتوزومال (انگلیسی است. شروع بلوغ دیابت جوان- دیابت بزرگسالان جوان)، که با نقض مشخص می شود، اما عدم وجود ترشح انسولین با حساسیت طبیعی به بافت های محیطی نیست.
جدول. 7.1طبقه بندی دیابت
به طور کلی به ندرت یافت می شود نقص ژنتیک انسولینجهش گیرنده انسولین متصل شده (leprechaunism، سندرم قفسه سینه). سی دی به طور طبیعی با توسعه بیماری های عصبی از پانکراس،منجر به تخریب سلول های بتا (پانکراتیت، پانکراتکتومی، فیبروز کیستیک، هموکوروماتوز)، و همچنین تحت تعدادی از بیماری های غدد درون ریز، که در آن محصولات بیش از حد از هورمون های تداوم (Acromegaly، سندرم کوشینگ) رخ می دهد. داروهای دارویی و مواد شیمیایی(واکر، پنتامیدین، اسید نیکوتین، دیازاکسید، و غیره) به ندرت علت SD است، اما ممکن است به تظاهرات و عدم توانایی بیماری در افراد مبتلا به مقاومت به انسولین کمک کند. ردیف بیماری های عفونی(سرخجه، سیتومگالی، کوک ها و عفونت آدنوویال) ممکن است با تخریب سلول های بتا همراه باشد، در حالی که اکثر بیماران نشانگرهای ایمنی زایی SD-1 را تعریف می کنند. به اشکال نادر از دیابت متداول ایمونوsD، در حال توسعه در بیماران مبتلا به STIFF-RNAN-STIFF-RNAN (بیماری نورولوژیک اتوایمیون)، و همچنین دیابت به علت اثرات آنتیبادی ها به گیرنده های انسولین. انواع مختلف SD با افزایش فرکانس در آن یافت می شود
بسیاری از سندرم های ژنتیکی، به ویژه در سندرم های پایین، Klinfelter، Turner، Tungsten، Prader-Willie و تعدادی از دیگران.
7.2. جنبه های بالینی متابولیسم کربوهیدرات
انسولیناین سنتز شده و ترشح شده توسط β- سلول های لانگرهان از پانکراس (PJZ). علاوه بر این، جزایر Langerhans اسرار گلوکاگون (سلول های α)، سموتوستاتین (سلول های δ) و پلیپپتید پانکراس (سلول PP) را ترشح می کند. هورمون های سلول های جزیره با یکدیگر ارتباط برقرار می کنند: گلوکاگون به طور معمول ترشح انسولین را تحریک می کند و somatostatin ترشح انسولین و گلوکاگون را سرکوب می کند. مولکول انسولین شامل دو زنجیره پلی پپتید (A-chain - 21 آمینو اسید؛ در زنجیره - 30 آمینو اسید) (شکل 7.1). سنتز انسولین شروع می شود با تشکیل پره پروینولین، که پروتئاز را به آموزش تقسیم می کند proinsulinدر گرانول های ترشحی دستگاه، گلگی، پینلین به انسولین تقسیم می شود C-peptide،که در فرآیند اگزوسیتوز به خون منتقل می شود (شکل 7.2).
محرک اصلی ترشح انسولین گلوکز است. انتشار انسولین در پاسخ به افزایش گلوکز خون دو مرحلهای(شکل 7.3). اولین یا فاز حاد چند دقیقه طول می کشد و با انتشار انباشت همراه است
 شکل. 7.1نمودار ساختار اولیه مولکول انسولین
شکل. 7.1نمودار ساختار اولیه مولکول انسولین
 شکل. 7.2.طرح بیوسنتز انسولین
شکل. 7.2.طرح بیوسنتز انسولین
shegne در یک انسولین سلول β- سلول در دوره بین وعده های غذایی. مرحله دوم ادامه می یابد تا سطح گلیسمی به بازرگان نرمال برسد (3.3-5.5 mmol / l). به طور مشابه، سلول های بتا بر آماده سازی سولفونیل اوره تاثیر می گذارد.
با توجه به سیستم انسولین پورتال می رسد کبد- ارگان اصلی اصلی آن. گیرنده های کبدی نیمی از هورمون ترشح می شوند. نیمه دیگر، سقوط به جریان خون سیستمیک، به عضلات و بافت های چربی می رسد. اکثر انسولین (80٪) تحت فروپاشی پروتئولیتیک در کبد قرار می گیرند، بقیه در کلیه ها قرار دارند و تنها مقدار جزئی به طور مستقیم با سلول های عضلانی و چربی متابولیزه می شود. Norma PJZ.
 شکل. 7.3.آزادی انسولین دو فاز تحت تاثیر گلوکز
شکل. 7.3.آزادی انسولین دو فاز تحت تاثیر گلوکز
مرد بالغ 35-50 واحد در روز را ترشح می کند که 0.6-1.2 واحد در هر کیلوگرم وزن بدن است. این ترشح به تغذیه و بازال تقسیم می شود. ترشح غذاانسولین CO از سطح بالابر پس از زایمان از گلوکز استفاده می کند. با توجه به آن، خنثی سازی غذاهای hyperglycimizing تضمین شده است. مقدار انسولین غذایی تقریبا مربوط به تعداد کربوهیدرات های گرفته شده است - حدود 1-2.5
10-12 گرم کربوهیدرات (1 واحد نان - هی). انسولین ترشح پایهسطح مطلوب گلیسمی و آنابولیسم را در فواصل بین غذا و در طول خواب فراهم می کند. انسولین پایه با سرعت حدود 1 UN / H ترشح می شود، با تمرین طولانی مدت یا گرسنگی طولانی، به طور قابل توجهی کاهش می یابد. انسولین غذایی حداقل 50 تا 70 درصد تولید روزانه انسولین را تشکیل می دهد (شکل 7.4).
ترشح انسولین نه تنها به غذا، بلکه همچنین روزانه
 شکل. 7 .4.
تولید روزانه انسولین نورما
شکل. 7 .4.
تولید روزانه انسولین نورما
نوسانات:نیاز به انسولین در ساعتهای صبح زود افزایش می یابد و در آینده به تدریج در طول روز سقوط می کند. بنابراین، کفش های ورزشی 2.0-2.5 برای صبحانه در 1 HEB، برای ناهار - 1.0-1.5 واحد، و برای شام - 1.0 واحد، ترشح می شوند. یکی از دلایل چنین تغییری در حساسیت به انسولین، سطح بالایی از هورمون های کنونی (عمدتا کورتیزول) در ساعت های صبح است که به تدریج در ابتدای شب به حداقل می رسد.
پایه ای اثرات فیزیولوژیکی انسولینتحریک انتقال گلوکز از طریق غشاهای بافت وابسته به انسولین وجود دارد. جسد اصلی انسولین کبد، بافت چربی و عضله است. به بافت های وابسته به انسولین، جریان گلوکز که به اثرات انسولین بستگی ندارد، عمدتا شامل سیستم عصبی مرکزی و محیطی، اندوتلیوم عروق خونی، سلول های خونی، و غیره است. انسولین باعث سنتز گلیکوژن در کبد می شود و عضلات، سنتز چربی در بافت کبد و چربی، پروتئین سنتز در کبد، عضلات و سایر اندام ها. تمام این تغییرات به استفاده از گلوکز هدایت می شود که منجر به کاهش سطح آن در خون می شود. آنتاگونیست انسولین فیزیولوژیکی است گلوکگونکه باعث تحریک بسیج گلیکوژن و چربی ها از انبار می شود؛ به طور معمول، سطح گلوکاگون محصولات انسولین متقابل را تغییر می دهد.
اثرات بیولوژیکی انسولین توسط گیرندهکه در سلول های هدف قرار دارند. گیرنده انسولین یک گلیکوپروتئین است که شامل چهار واحد است. با سطح بالایی از انسولین در خون، تعداد گیرنده های آن بر اصل مقررات پایین تر کاهش می یابد، که با کاهش حساسیت سلولی به انسولین همراه است. پس از اتصال انسولین با گیرنده سلولی، این مجموعه درون سلول قرار می گیرد. بعد از آن در داخل عضله و سلول چربی، انسولین باعث بسیج حنجرهای داخل سلولی می شود که حاوی آن هستند نوار نقاله گلوکزگلوت 4 در نتیجه، vesicles به سطح سلولی منتقل می شود، جایی که Glut-4 عملکرد ورودی برای گلوکز را انجام می دهد. اثر مشابهی بر GLUT-4 دارای اعمال فیزیکی است.
7.3. تشخیص آزمایشگاه و معیارهای جبران دیابت
تشخیص آزمایشگاهی SD بر اساس تعیین سطح قند خون است، در حالی که معیارهای تشخیص برای همه متحد هستند
انواع و انواع SD (جدول 7.2). داده های سایر مطالعات آزمایشگاهی (سطح گلوکوزوریا، تعریف سطح هموگلوبین گلیکواد) نباید مورد استفاده قرار گیرد تا تشخیص تشخیص را بررسی کند. تشخیص SD را می توان بر اساس تشخیص دو بار از یکی از سه معیار:
1. با علائم SD واضح (پلیوریا، پلیدیپسی) و سطح گلوکز در خون مویرگی جامد، بیش از 11.1 میلی مول در لیتر، صرف نظر از زمان روز و قبل از غذا.
2. در سطح گلوکز در یک خون مویرگی جامد، معده خالی بیش از 6.1 mmol / l.
3. در سطح گلوکز در خون مویرگی جامد 2 ساعت پس از دریافت 75 گرم گلوکز (آزمون گلوکز خوراکی) بیش از 11.1 میلی مول در لیتر.
جدول. 7.2.معیارهای تشخیص دیابت
 مهمترین و مهمترین آزمون در تشخیص سی دی این است که تعیین سطح گلیسمی بر روی معده خالی (حداقل 8 ساعت روزه). در فدراسیون روسیه، سطح گلیسمی معمولا در خون جامد تخمین زده می شود. بسیاری از کشورها به طور گسترده ای برای تعیین سطح گلوکز استفاده می شوند
مهمترین و مهمترین آزمون در تشخیص سی دی این است که تعیین سطح گلیسمی بر روی معده خالی (حداقل 8 ساعت روزه). در فدراسیون روسیه، سطح گلیسمی معمولا در خون جامد تخمین زده می شود. بسیاری از کشورها به طور گسترده ای برای تعیین سطح گلوکز استفاده می شوند
در پلاسمای خون. تست گلوکز خوراکی(OGTT؛ تعیین سطح گلوکز 2 ساعت پس از تزریق داخل 75 گرم گلوکز حل شده در آب) در این زمینه، ارزش کمتر وجود دارد. با این وجود، بر اساس OGTT تشخیص داده می شود نقض تحمل گلوکز(NTG). Nth تشخیص داده می شود اگر سطح گلیقیم خون مویرگی جامد بیش از 6.1 mmol / l باشد، و 2 ساعت پس از بار، گلوکز بالاتر از 7.8 mmol / l است، اما کمتر از 11.1 mmol / l است. یکی دیگر از تجسم مبادلات کربوهیدرات است گلیسمی نقض شده در یک فروشگاه خالی(NGN). دومی ثابت شده است اگر سطح گلیسمی خون مویرگی جامد در معده خالی در محدوده 5.6-6.0 mmol / L و 2 ساعت پس از بار با گلوکز کمتر از 7.8 mmol / l) باشد. NTG و NGNT در حال حاضر توسط این اصطلاح متحد هستند پیشبریاز آنجایی که هر دو دسته از بیماران خطر بسیار بالایی از تظاهرات SD و توسعه ماکرونگتیوپاتی دیابتی دارند.
برای تشخیص SD، سطح گلیسمی باید با روش های استاندارد آزمایشگاهی تعیین شود. در تفسیر شاخص های گلیسمی، باید در نظر گرفته شود که سطح معده خالی گلوکز در خون جامد وریدی به سطح آن در یک مویرگی جامد مربوط می شود. پس از دریافت غذا یا OGTT، سطح آن در خون وریدی حدود 1.1 mmol / l کمتر از مویرگی است. محتوای گلوکز در پلاسما حدود 0.84 mmol / l بالاتر از خون جامد است. به منظور ارزیابی جبران خسارت و کفایت CD درمانی، سطح گلیسمی در خون مویرگی با استفاده از قابل حمل تخمین زده می شود گلوکومتربیماران خود، بستگان آنها یا پرسنل پزشکی.
با هر نوع دیابت، و همچنین با بار قابل توجهی از گلوکز می تواند توسعه یابد گلوکوزوریکدام یک از نتایج بیش از حد آستانه جذب گلوکز از ادرار اولیه است. آستانه جذب گلوکز به طور قابل توجهی متفاوت است (≈ 9-10 mmol / l). به عنوان یک شاخص جداگانه گلوکوزوریا برای تشخیص SD نباید استفاده شود. به طور معمول، به استثنای موارد بارگیری مواد غذایی قابل توجه کربوهیدرات های تصفیه شده، گلوکوزوریا یافت نشد.
محصولات پارچه کتون(استون، استون، acetoacetate، β-hydroxybutyrate) به طور قابل توجهی در کمبود انسولین مطلق تشدید می شود. هنگامی که Decompensation، SD-1 ممکن است تلفظ کند کتونوری(به بررسی نوارهای تست، که در ادرار کاهش می یابد). آسان (ردیابی) Ketonuria را می توان در افراد سالم با گرسنگی و رژیم غذایی تخمیر تعیین کرد.
شاخصی مهم آزمایشگاهی که برای تشخیص افتراقی انواع SD استفاده می شود، و همچنین شناسایی تشکیل کمبود انسولین در بیماران مبتلا به SD-2، سطح آن است پپتید Cاز لحاظ سطح پپتید C در خون، می توان از توانایی قرار دادن بتا سلول های PJZ قضاوت کرد. دومی تولید پروینولین، که از آن پپتید C قبل از ترشح شکسته می شود، که در مقادیر مشابه با انسولین به خون می رسد. انسولین 50٪ در کبد تماس گرفته و نیمه عمر در خون محیطی حدود 4 دقیقه دارد. C-peptide از کبد جریان خون حذف نمی شود و دارای نیمه عمر در خون حدود 30 دقیقه است. علاوه بر این، آن را با گیرنده های سلولی در حاشیه ارتباط ندارد. بنابراین، تعریف سطح C-peptide یک آزمون قابل اعتماد تر برای تخمین عملکرد دستگاه های مبهم است. سطح پپتید C بیشتر به طور اطلاعاتی در برابر پس زمینه نمونه های تحریک (پس از خوردن یا مصرف گلوکاگون) مورد بررسی قرار می گیرد. آزمون غیر قابل استفاده است اگر آن را در برابر پس زمینه از عدم اندازه گیری شدید SD انجام می شود، از آنجایی که هیپرگلیسمی تلفظ شده اثر سمی بر روی سلول های بتا (گلوکوزوتوکسیسیت) دارد. درمان انسولین برای چندین روز قبل به نتایج آزمون تاثیر نمی گذارد.
اصلی هدف از درمانهر نوع CD برای جلوگیری از عوارض دیررس آن است که می تواند در برابر پس زمینه جبران ضخامت آن بر روی تعدادی از پارامترها (جدول 7.3) به دست آید. معیار اصلی کیفیت جبران خسارت برای متابولیسم کربوهیدرات تحت CD سطح است هموگلوبین گلیسیل شده (گلیکوزیل شده) (HbA1c).دومی هموگلوبین است، ناشناخته با گلوکز همراه است. در اریتروسیت های گلوکز به طور مستقل از انسولین می آید و گلیکوزیل شدن هموگلوبین یک فرآیند غیرقابل برگشت است و درجه آن به طور مستقیم با غلظت گلوکز متناسب است که با آن 120 روز از وجود آن تماس گرفته شده است. بخش کوچکی از هموگلوبین گلیکوزیل شده و طبیعی است؛ با سی دی، می توان آن را به طور قابل توجهی افزایش داد. سطح HBA1C، در مقایسه با سطح گلوکز، که به طور مداوم در حال تغییر است، به طور یکپارچه نشان دهنده گلیسمی در طول 3-4 ماه گذشته است. این با این فاصله است که سطح HBA1C برای ارزیابی جبران SD توصیه می شود.
هیپرگلیسمی مزمن دور از تنها عامل خطر برای توسعه و پیشرفت عوارض دیررس SD است. مربوط به ارزیابی SD جبرانبر اساس مجموعه
روش های تحقیقات آزمایشگاهی و ابزار (جدول 7.3). علاوه بر شاخص هایی که مشخصه شرایط متابولیسم کربوهیدرات را مشخص می کنند، سطح فشار خون و طیف لیپید خون مهمترین آنها هستند.
جدول. 7.3.معیارهای جبران دیابت شکر
 علاوه بر معیارهای جبران بالا، یک رویکرد فردی در هنگام برنامه ریزی اهداف درمان CD مورد نیاز است. احتمال توسعه و پیشرفت عوارض دیررس SD (به ویژه میکروآنژیوپاتی) با افزایش طول مدت بیماری افزایش می یابد. بنابراین، اگر در کودکان و بیماران جوان، طول دیابت در آینده می تواند به چند دهه برسد، لازم است به دست آوردن شاخص های بهینه گلیسمی، و سپس در بیمارانی که در سالخوردگان و پیری آشکار می شوند، جبران خسارت شدید به طور قابل توجهی بهبود خطر هیپوگلیسمی، همیشه مناسب نیست.
علاوه بر معیارهای جبران بالا، یک رویکرد فردی در هنگام برنامه ریزی اهداف درمان CD مورد نیاز است. احتمال توسعه و پیشرفت عوارض دیررس SD (به ویژه میکروآنژیوپاتی) با افزایش طول مدت بیماری افزایش می یابد. بنابراین، اگر در کودکان و بیماران جوان، طول دیابت در آینده می تواند به چند دهه برسد، لازم است به دست آوردن شاخص های بهینه گلیسمی، و سپس در بیمارانی که در سالخوردگان و پیری آشکار می شوند، جبران خسارت شدید به طور قابل توجهی بهبود خطر هیپوگلیسمی، همیشه مناسب نیست.
7.4 آماده سازی انسولین و درمان انسولین
آماده سازی انسولین برای بیماران مبتلا به SD-1 حیاتی است؛ علاوه بر این، آنها تا 40٪ بیماران مبتلا به SD-2 دریافت می کنند. به رایج نشانه های انتصاب درمان انسولین در SD،بسیاری از آنها در واقع یکدیگر را پوشش می دهند عبارتند از:
1. دیابت نوع 1
2. panketectomy
3. کما کتوسیدیوتیک و هیپراتوماتیک
4. با دیابت نوع 2:
نشانه های صریح کمبود انسولین، مانند پیشرفت وزن بدن و کتوز، هیپرگلیسمی بیان شده؛
مداخلات جراحی بزرگ؛
عوارض حاد کلان پیچیده (سکته مغزی، انفارکتوس میوکارد، گانگرن، و غیره) و بیماری های شدید عفونی، همراه با عدم تمرکز متابولیسم کربوهیدرات؛
سطح گلیسمی معده خالی بیش از 15-18 mmol / l است؛
فقدان پرداخت جبران خسارت، علیرغم تجویز دوز روزانه داروهای مختلفی از داروهای بازتابنده تبلت؛
مراحل اواخر عوارض دیررس SD (پلی اوروپاتی شدید و رتینوپاتی، نارسایی مزمن کلیوی).
5. ناتوانی در جبران دیابت حاملگی با استفاده از رژیم غذایی و درمان.
توسط مبداآماده سازی انسولین را می توان به سه گروه تقسیم کرد:
حیوانات انسولین (گوشت خوک)؛
انسولین انسانی (مهندسی نیمه مصنوعی، مهندسی ژنتیک)؛
آنالوگ های انسولین (Lizpro، Aspart، Glargin، Demide).
پیشرفت فن آوری برای تولید انسولین انسانی منجر به استفاده از انسولین خوک(متفاوت از یک اسید آمینه انسان) به تازگی به طور قابل توجهی کاهش یافته است. انسولین گوشت خوک را می توان برای تولید انسولین انسانی استفاده کرد روش نیمه مصنوعیکه به معنای جایگزینی یک اسید آمینه متفاوت در مولکول آن است. بالاترین کیفیت متفاوت است مهندسی ژنتیکیانسولین انسانی برای به دست آوردن آنها، سایت ژنوم انسان مسئول سنتز انسولین همراه با ژنوم است e.coliیا فرهنگ مخمر، به عنوان یک نتیجه از آن شروع به تولید انسولین انسانی. موجود انسولین آنالوگبا کمک جایگزینی از اسیدهای آمینه های مختلف، هدف از دست رفتن داروها با داروهای داده شده و مطلوب، به دست آمد. بنابراین، انسولین Lizpro (Humalog) آنالوگ است
انسولین از عمل فوق العاده فوق العاده، در حالی که اثر ساکشی آن پس از 15 دقیقه پس از تزریق رشد می کند. برعکس، یک آنالوگ از انسولین گلورژین (لانتوس)، برعکس، با یک اقدام طولانی که در طول روز ادامه می یابد، مشخص می شود، در حالی که ویژگی سینتیک مواد مخدر کمبود قله های پلاسما است. بیشتر از آماده سازی انسولین در حال حاضر استفاده شده و آنالوگ های آن تولید می شود تمرکز100 U / میلی لیتر توسط مدت زمان عملانسولین به 4 گروه اصلی تقسیم می شوند (جدول 7.4):
جدول. 7.4فارماکوکینتیک مواد مخدر و آنالوگ انسولین
 1.
عمل UltraShort (LizPro، Aspart).
1.
عمل UltraShort (LizPro، Aspart).
2. اقدام کوتاه (انسولین ساده انسان).
3. میانگین مدت عمل (انسولین ها در هگارتر پروتامین خنثی).
4. اقدام بلند مدت (Glagragin، Detech).
5. مخلوط انسولین مدت زمان های مختلف (Novomix-30، Humulin-MH، Humalog Mix-25).
آماده سازی عمل فوق العاده[LizPro (Humalog)، آسپارت (Novorad)] آنالوگ انسولین است. مزایای آنها توسعه سریع اثر اثر قند پس از تزریق (پس از 15 دقیقه)، که به شما اجازه می دهد تا بلافاصله قبل از غذا و یا حتی بلافاصله پس از غذا، تزریق کنید، و همچنین مدت کوتاهی از عمل (کمتر از 3 ساعت) ، که خطر ابتلا به هیپوگلیسمی را کاهش می دهد. آماده سازی اقدام کوتاه(انسولین ساده، انسولین منظم) یک محلول حاوی انسولین در غلظت 100 واحد / میلی لیتر است. تزریق انسولین ساده 30 دقیقه قبل از غذا ساخته شده است؛ مدت زمان عمل حدود 4-6 ساعت است. آماده سازی فوق العاده پیچ و عمل کوتاه می تواند به صورت زیر جلدی، عضلانی و داخل وریدی تزریق شود.
در میان مواد مخدر میانگین مدت اقداماغلب آماده سازی ها در Hageorn پروتامین خنثی (NPH) استفاده می شود. NPH پروتئینی است که انسولین adsorb ناشناخته است و مکش خود را از انبار زیر جلدی کاهش می دهد. مدت موثر عمل انسولین NPH معمولا حدود 12 ساعت است؛ آنها فقط به صورت زیر جلدی وارد می شوند. انسولین NPH تعلیق است، در ارتباط با آن، در مقایسه با انسولین ساده در ویال، آن گل آلود است، و با ایستادگی طولانی، تعلیق وجود دارد، که باید قبل از تزریق کاملا مخلوط شود. انسولین NPH بر خلاف سایر آماده سازی اقدامات طولانی مدت می تواند با یک انسولین کوتاه مدت (انسولین ساده) مخلوط شود، در حالی که فارماکوکینتیک اجزای مخلوط تغییر نخواهد کرد، زیرا NPH مقدار اضافی انسولین ساده را متصل نخواهد کرد (شکل 7.5). علاوه بر این، پروتامین برای آماده سازی مخلوط های استاندارد آنالوگ های انسولین (Novomix-30، Humalog-Mix-25) استفاده می شود.
در میان داروهای اقدام طولانی مدت در حال حاضر به طور فعال از آنالوگ انسولین استفاده می کنند گلور(لانتوس) و دیمر(Leewemir). ویژگی مطلوب فارماکوکینتیک این داروها این است که در مقایسه با انسولین NPC، آنها جریان یکنواخت و طولانی مدت دارو را از انبار زیر جلدی ارائه می دهند. در این راستا، گلورژین را می توان تنها یک بار در روز منصوب کرد، در حالی که تقریبا هیچ وقت صرف نظر از زمان روز نیست.
 شکل. 7.5.Pharmacokokinetics از آماده سازی انسولین مختلف:
شکل. 7.5.Pharmacokokinetics از آماده سازی انسولین مختلف:
الف) مونو کامپوننت؛ ب) مخلوط انسولین استاندارد
علاوه بر انسولین داروهای تکمیلی، عمل بالینی به طور گسترده ای مورد استفاده قرار می گیرد مخلوط های استانداردبه عنوان یک قاعده، ما در مورد مخلوط های انسولین کوتاه یا فوق العاده با انسولین مدت زمان عمل صحبت می کنیم. به عنوان مثال، دارو "Humulin-MW" شامل یک بطری 30٪ از انسولین ساده و 70٪ انسولین NPH؛ این دارو "Novomiks-30" شامل 30٪ انسولین آسپارت و 70٪ از تعلیق پروتامین کریستال انسولین آسپارت؛ این دارو "Humalog-mix-25" حاوی 25٪ انسولین LysPro و 75٪ پروتئین تعلیق اجاره انسولین است. مزیت - فایده - سود - منفعت
مخلوط انسولین استاندارد جایگزینی دو تزریق یک و چند دقت زیادی از اجزای مخلوط است. ناکارآمدی عدم امکان دوزهای فردی از اجزای فردی مخلوط است. این تعیین ترجیح استفاده از مخلوط های انسولین استاندارد برای درمان CD-2 یا با به اصطلاح است درمان انسولین سنتی(انتصاب دوزهای ثابت انسولین)، در حالی که برای درمان انسولین شدید(انتخاب دوز انعطاف پذیر بسته به شاخص های گلیسمی و مقدار کربوهیدرات در غذا) به استفاده از داروهای تکمیلی ترجیح داده می شود.
کلید درمان موفق به انسولین موفقیت آمیز است تکنیک های تزریقچندین راه برای معرفی انسولین وجود دارد. ساده ترین و ساده ترین روش قابل اعتماد - تزریق با انسولین سرنگ.یک روش راحت تر برای معرفی انسولین تزریق استفاده می شود دستگیره های سرنگکدام یک از دستگاه های ترکیبی حاوی مخزن انسولین (کارتریج)، یک سیستم دوز و سوزن انژکتور است.
برای حمایت از درمان (زمانی که آن را به تقلید از SD یا در مورد دولت های بحرانی می آید)، انسولین به صورت زیر جلدی معرفی می شود. تزریق انسولین یک اقدام کوتاه توصیه می شود که به بافت چربی زیر جلدی شکم، انسولین عمل طولانی مدت - به فیبر لگن یا شانه (شکل 7.6 a) انجام دهید. تزریق به داخل بافت زیر جلدی از طریق پوست به طور گسترده ای فشرده شده در زاویه 45 درجه (شکل 7.6 B) ساخته شده است. بیمار باید یک تغییر روزانه سایت های تزریق انسولین را در همان منطقه به منظور جلوگیری از توسعه لیپودیزستروف توصیه کند.
به عوامل موثر بر سرعت جذب انسولیناز انبار زیر جلدی، دوز انسولین باید نسبت داده شود (افزایش دوز طول جذب را افزایش می دهد)، محل تزریق (جذب سریعتر از فیبر شکمی)، دمای محیط (گرمایش و ماساژ محل تزریق، جذب جذب را افزایش می دهد )
یک روش مدیریت پیچیده تر که، با این حال، در بسیاری از بیماران به شما اجازه می دهد تا نتایج خوب درمان را به دست آورید، استفاده از آن است تلگراف انسولینیا سیستم هایی برای تزریق انسولین زیر جلدی مداوم. تلگراف یک دستگاه قابل حمل است که متشکل از یک کامپیوتر است که حالت عرضه انسولین را تنظیم می کند، و همچنین یک سیستم عرضه انسولین، بر روی یک کاتتر و سوزن مینیاتوری به زیر جلدی انجام می شود
 شکل. 7.6.تزریق تزریق: الف) مکان های تزریق معمولی؛ ب) موقعیت سوزن سرنگ انسولین در طی تزریق
شکل. 7.6.تزریق تزریق: الف) مکان های تزریق معمولی؛ ب) موقعیت سوزن سرنگ انسولین در طی تزریق
بافت چربی. با کمک تلگراف، معرفی یکپارچه مداوم یک انسولین کوتاه یا فوق العاده (میزان حدود 0.5-1 در ساعت) انجام می شود و قبل از مصرف غذا، بسته به محتوای کربوهیدرات ها و سطح گلیسمی، بیمار دوز بولوس لازم را از همان انسولین یک اقدام کوتاه را معرفی می کند. مزیت درمان انسولین با کمک تلگراف، معرفی یک انسولین یک اقدام کوتاه (یا حتی ultrashort) است که به خودی خود بیشتر فیزیولوژیک است، زیرا جذب آماده سازی انسولین طولانی مدت در معرض نوسانات بزرگ قرار دارد؛ در این راستا، معرفی مداوم یک انسولین کوتاه کوتاه، به نظر می رسد یک فرآیند قابل کنترل است. ضرر درمانی درمان انسولین با کمک تلگراف، نیاز به دستگاه حمل ثابت، و همچنین پایه طولانی مدت یک سوزن تزریق در بافت زیر جلدی است که نیاز به کنترل دوره ای بر روند عرضه انسولین دارد. درمان انسولین با کمک یک تلگراف به طور عمده به بیماران مبتلا به SD-1 نشان داده شده است، که آماده تکنیک تعمیر و نگهداری آن هستند. به خصوص در این رابطه، شما باید به بیماران مبتلا به پدیده ای از "صبح روز صبح"، و همچنین برای زنان باردار و برنامه ریزی بیماران بارداری با SD-1 و پاریس توجه کنید
با یک شیوه اختلال زندگی (امکان یک حالت قدرت انعطاف پذیر تر).
7.5. دیابت نوع 1
SD-1 - Organship خاص خود ایمنیاین بیماری منجر به تخریب سلول های بتا تولید انسولین از جزایر PJZ، که توسط کمبود انسولین مطلق ظاهر می شود. در برخی موارد، بیماران مبتلا به SD-1 صریح نشانگرهای ضایعه اتوایمیون از سلول های بتا را ندارند (SD-1 Idiopathic).
اتیولوژی
SD-1 یک بیماری با استعداد ارثی است، اما سهم آن در توسعه بیماری کوچک است (توسعه آن را در حدود 1 / ثانیه تعیین می کند). همبستگی دوقلوهای تک نفره در SD-1 تنها 36٪ است. احتمال توسعه SD-1 در یک کودک مبتلا به مادر بیمار 1-2٪، پدر - 3-6٪، برادر یا خواهر - 6٪ است. برخی از یا چند نشانگر هومورال از ضایعات اتوایمیون از سلول های بتا، که آنتی بادی ها شامل آنتی بادی های PJZ، آنتی بادی های گلوتامات دکربوکسیلاز (GAD65) و آنتی بادی های تریروزین فسفاتاز (IA-2 و IA-2 و IA-2β) در 85-90٪ یافت می شود از بیماران.. با این وجود، عوامل ایمنی سلولی به تخریب سلول های β متصل می شوند. SD-1 با Haplotypes HLA مانند dqaو DQB،در همان زمان به تنهایی آلل hla-dr / dqممکن است به توسعه بیماری منجر شود، در حالی که دیگران اعتراض می کنند. با افزایش فراوانی SD-1، همراه با سایر غدد درون ریز اتوایمیون (تیروئیدیت اتوایمیون، بیماری آدیسون) و بیماری های غیر الکل مانند آلوپسی، ویتیلیگو، بیماری تاج، بیماری های روماتیسمی (جدول 7.5).
پاتوژنز
SD-1 در طول تخریب یک فرایند اتوایمیون 80-90٪ سلول های بتا آشکار می شود. سرعت و شدت این فرآیند می تواند به طور قابل توجهی متفاوت باشد. اغلب جریان معمولیبیماری ها در کودکان و نوجوانان این روند به سرعت به سرعت ادامه می یابد و پس از آن یک تظاهرات خشونت آمیز بیماری، که در آن ظهور اولین علائم بالینی به توسعه کتواسییدوز (تا کاستواسیدوتیک) می تواند تنها چند هفته طول بکشد.
جدول. 7.5.دیابت نوع 1
 ادامه جدول 7.5.
ادامه جدول 7.5.
 به طور مداوم، به طور قابل توجهی بیشتر موارد نادر، به عنوان یک قاعده، در بزرگسالان بیش از 40 سال، بیماری می تواند جریان پنهان باشد (دیابت خودآموزی پنهان بزرگسالان - لادا)در همان زمان، در اولین بیماری، چنین بیماران اغلب تشخیص SD-2 را ایجاد می کند و طی طی چندین سال، جبران خسارت CD را می توان با قرار ملاقات داروهای سولفونیلوروری به دست آورد. اما در آینده، معمولا 3 سال بعد، علائم کمبود انسولین مطلق (کاهش وزن، کتونوری، هیپرگلیسمی بیان شده، به رغم پذیرش داروهای ساحلی تبلت، وجود دارد.
به طور مداوم، به طور قابل توجهی بیشتر موارد نادر، به عنوان یک قاعده، در بزرگسالان بیش از 40 سال، بیماری می تواند جریان پنهان باشد (دیابت خودآموزی پنهان بزرگسالان - لادا)در همان زمان، در اولین بیماری، چنین بیماران اغلب تشخیص SD-2 را ایجاد می کند و طی طی چندین سال، جبران خسارت CD را می توان با قرار ملاقات داروهای سولفونیلوروری به دست آورد. اما در آینده، معمولا 3 سال بعد، علائم کمبود انسولین مطلق (کاهش وزن، کتونوری، هیپرگلیسمی بیان شده، به رغم پذیرش داروهای ساحلی تبلت، وجود دارد.
اساس پاتوژنز SD-1، به عنوان نشان داده شده، کمبود انسولین مطلق است. عدم امکان ورود گلوکز به پارچه های وابسته به انسولین (چربی و عضلانی) منجر به نارسایی انرژی می شود که منجر به افزایش لیپولیز و پروتئولیز می شود که از دست دادن وزن بدن مرتبط است. افزایش سطح گلیسمی باعث افزایش هیپروسومولاری می شود که همراه با دیورز اسمزی و کمبود آب بدن همراه است. در شرایط کمبود انسولین و نارسایی انرژی، محصولات هورمون پیونر در حال توسعه هستند (گلوکاگون، کورتیزول، هورمون رشد)، که، با وجود افزایش گلیسمی، تحریک گلوکگنز را تعیین می کند. افزایش لیپولیز در بافت چربی منجر به افزایش قابل توجهی در غلظت اسیدهای چرب آزاد می شود. با کمبود انسولین، توانایی لیپوسینتتیک کبد سرکوب می شود و
اسیدهای چرب در Ketogenesis شروع به روشن شدن می کنند. انباشت اجسام کتون منجر به توسعه کتوز دیابتی می شود و در آینده - کتواسییدوز. با افزایش پیشرونده کمبود آب و اسیدوز، یک حالت کموتوز در حال توسعه است (به بند 7.7.1 مراجعه کنید)، که در غیاب درمان انسولین و ریویتر، ناگزیر با مرگ به پایان می رسد.
همهگیرشناسی
در SD-1، حدود 1.5-2٪ از تمام موارد دیابت وجود دارد و این رقم نسبی به دلیل رشد سریع بروز SD-2 ادامه خواهد یافت. خطر ابتلا به SD-1 در طول عمر نماینده نژاد سفید حدود 0.4٪ است. بروز SD-1 به میزان 3 درصد افزایش می یابد: 1.5 درصد به علت موارد جدید و 1.5 درصد دیگر به دلیل افزایش امید به زندگی بیماران، 1.5 درصد است. شیوع SD-1 بسته به ترکیب قومی جمعیت متفاوت است. برای سال 2000، آن را به میزان 0.02٪ در آفریقا، 0.1٪ در جنوب آسیا، و همچنین در آمریکای جنوبی و آمریکای جنوبی و 0.2٪ در اروپا و آمریکای شمالی بود. بیشترین میزان بروز SD-1 در فنلاند و سوئد (30-35 مورد در هر 100 هزار جمعیت در سال) و پایین ترین در ژاپن، چین و کره (به ترتیب 0.5-2.0 مورد) است. پیک مربوط به سن تظاهرات SD-1 مربوط به حدود 10 تا 13 سال است. در اکثریت قریب به اتفاق موارد، SD-1 تا 40 سال ظاهر می شود.
تظاهرات بالینی
که در موارد معمولیبه خصوص در کودکان و نوجوانان، SD-1 یک تصویر بالینی روشن است که برای چند ماه یا حتی هفته ها توسعه می یابد. تظاهرات SD-1 می تواند عفونی و سایر بیماری های همزمان را تحریک کند. مشخصه مشترک برای همه انواع علائم SD،مرتبط با هیپرگلیسمی: پلیدیپسی، پلیوریا، خارش پوست، اما در SD-1 آنها بسیار تلفظ شده اند. بنابراین، در طول روز، بیماران می توانند تا 5-10 لیتر مایع مصرف کنند و استخراج کنند. خاصبرای SD-1، علامت، که به علت کمبود مطلق انسولین است، کاهش وزن است و به مدت 1-2 ماه به 10-15 کیلوگرم رسید. این با ضعف کلی و عضلانی شدید، کاهش عملکرد، خواب آلودگی مشخص می شود. در ابتدای بیماری، برخی از بیماران ممکن است افزایش اشتها را افزایش دهند، که جایگزین بی اشتهایی به عنوان کتواسییدوز می شود. دومی با ظاهر بوی استون (یا بوی میوه) از دهان، توش مشخص می شود
nOTA، استفراغ، اغلب درد در معده (pseudoperitonite)، کم آبیاری شدید و به پایان می رسد با توسعه یک حالت کاماتوز (به بند 7.7.1 مراجعه کنید). در برخی موارد، اولین تظاهرات SD-1 در کودکان یک اختلال پیشرونده از آگاهی است تا به کما در برابر پس زمینه بیماری های همزمان، به عنوان یک قانون، آسیب شناسی عفونی یا حاد جراحی.
در موارد نسبتا نادر توسعه SD-1 در افراد بالای 35-40 سال (دیابت بالقوه اتوایمیون پنهان)این بیماری می تواند بسیار روشن باشد (پلیدیپسی متوسط \u200b\u200bو پلیوریا، کمبود کاهش وزن بدن) و حتی با شانس با تعریف معمول از سطح گلیسمی آشکار می شود. در این موارد، بیمار اغلب تشخیص داده های SD-2 را ایجاد می کند (TSP) تجویز می شود که برای برخی از زمان ها جبران پذیرش CD قابل قبول را ارائه می دهند. با این وجود، برای چندین سال (اغلب در طول سال)، بیمار به نظر می رسد علائم ناشی از کسری مطلق رو به رشد انسولین: کاهش وزن، عدم امکان حفظ گلیسمی طبیعی در پس زمینه TSP، کتوز، کتواسییدوز.
تشخیصی
با توجه به اینکه SD-1 دارای یک تصویر بالینی روشن است و همچنین یک بیماری نسبتا نادر است، تعریف غربالگری سطح گلیسمی به منظور تشخیص SD-1 نشان داده نمی شود. احتمال توسعه بیماری در نزدیکی نزدیکترین بستگان بیماران کم است، که همراه با کمبود روش های موثر پیشگیری اولیه اولیه، SD-1، عدم انطباق مطالعه نشانگرهای بیماری ایمونوژنیک را تعیین می کند. تشخیص SD-1 در اکثریت قریب به اتفاق بر اساس شناسایی هیپرگلیسمی قابل توجه در بیماران مبتلا به تظاهرات شدید بالینی کمبود مطلق انسولین است. OGTT برای هدف تشخیص SD-1 باید به ندرت انجام شود.
تشخیص های افتراقی
در موارد مشکوک (تشخیص هیپرگلیسمی متوسط \u200b\u200bدر غیاب تظاهرات بالینی صریح، تظاهرات در نسبتا سالمندان)، و همچنین به منظور تشخیص دیفرانسیل با سایر انواع SD، برای تعیین سطح استفاده می شود پپتید C(پایه و 2 ساعت پس از غذا). ارزش تشخیصی غیر مستقیم در موارد مشکوک ممکن است تعریف داشته باشد نشانگرهای ایمنیSD-1 - آنتی بادی های خوراکی
PJZ، به گلوتاماتدکربوکسیلاز (GAD65) و تیروزین فسفاتاز (IA-2 و IA-2β). تشخیص دیفرانسیل SD-1 و SD-2 در جدول ارائه شده است. 7.6.
جدول. 7.6.تشخیص دیفرانسیل و تفاوت بین SD-1 و SD-2
 رفتار
رفتار
درمان هر نوع CD بر اساس سه اصل اساسی است: درمان Sakharosyncing (با درمان SD-1 - انسولین)، رژیم غذایی و آموزش بیمار. انسولین درمانیدر SD-1 می پوشد تعویضهدف آن حداکثر تقلید از محصولات فیزیولوژیکی هورمون به منظور دستیابی به معیارهای جبران پذیرفته شده (جدول 7.3) است. به ترشح فیزیولوژیکی انسولین تقریبی است درمان انسولین شدید.نیاز به انسولین مربوط به آن است ترشح پایه،این توسط دو تزریق انسولین از مدت زمان متوسط \u200b\u200bعمل (در صبح و عصر) یا یک تزریق انسولین طولانی مدت (شکر) ارائه شده است. کل دوز بازال
لینا نباید بیش از نیمی از کل نیاز روزانه برای آماده سازی باشد. ترشح انسولین غذا یا بولوسآن را با تزریق انسولین یک عمل کوتاه یا فوق العاده قبل از مصرف هر وعده غذایی جایگزین کنید، در حالی که دوز آن بر اساس مقدار کربوهیدرات ها محاسبه می شود، که قرار است در طول مصرف مواد غذایی آینده و سطح موجود گرفته شود گلیسمی توسط بیمار مبتلا به گلوکومتر قبل از تزریق انسولین تعیین می شود (شکل 7.7).
تقریبی طرح درمان انسولین فشرده،که تقریبا هر روز متفاوت خواهد بود، می تواند به شرح زیر باشد. آنها از این واقعیت ادامه می دهند که نیاز روزانه انسولین حدود 0.5-0.7 واحد در هر کیلوگرم وزن بدن است (برای بیمار با وزن بدن 70 کیلوگرم در حدود 35-50 واحد). حدود 1 / S - 1/2 از این دوز، انسولین عمل طولانی مدت (20-25 واحد)، انسولین 1/2-2 / s از عمل کوتاه یا فوق العاده است. دوز NPH انسولین به 2 تزریق تقسیم می شود: در صبح 2 / s از دوز آن (12 واحد)، در شب - 1 / s (8-10 واحد).
هدف مرحله اولدرمان انسولین مهر و موم طبیعی بودن سطح گلوکز بر روی معده خالی است. دوز شبانه انسولین NPH معمولا در 22-23 ساعت، صبح همراه با تزریق یک انسولین کوتاه در مقابل صبحانه معرفی می شود. هنگام انتخاب یک دوز شبانه انسولین NPH، لازم است که امکان توسعه یک عدد را در نظر داشته باشید
 شکل. 7.7.طرح درمان انسولین شدید
شکل. 7.7.طرح درمان انسولین شدید
پدیده های کاملا معمولی. علت هیپرگلیسمی صبح ممکن است دوز کمی از اقدامات طولانی مدت انسولین باشد، زیرا صبح نیاز به انسولین به طور قابل توجهی افزایش می یابد (پدیده "صبحانه صبح").علاوه بر کمبود دوز، بیش از حد آن می تواند منجر به هیپرگلیسمی صبح شود. پدیده somoga(somogyi)، هیپرگلیسمی postgoglycemic. این پدیده با این واقعیت توضیح داده شده است که حداکثر حساسیت بافتی به انسولین بین 2 تا 4 ساعت شب مشخص شده است. در این زمان، سطح هورمون های پایه اصلی (کورتیزول، هورمون رشد، و غیره) طبیعی است. اگر دوز عصر انسولین اقدام طولانی مدت بیش از حد باشد، در این زمان توسعه می یابد هیپوگلیسمیبالینی، این می تواند خود را یک خواب بد با رویاهای کابوس، اقدامات ناخودآگاه در رویا، سردرد صبح و خرابی آشکار سازد. توسعه در این زمان هیپوگلیسمی باعث ایجاد جبران کننده جبران کننده جبرانی جبرانی و سایر هورمون های پیوندی می شود هیپرگلیسمی در ساعت صبح.اگر در این وضعیت، کاهش نمییابد، اما برای افزایش دوز انسولین طولانی مدت، سرمایه گذاری در شب، هیپوگلیسمی شب و هیپرگلیسمی صبح تشدید می شود که در نهایت می تواند به سندرم سندرم انسولین مزمن (سندرم Somoga) منجر شود ، که ترکیبی از چاقی با کمبود مزمن SD، هیپوگلیسمی مکرر و عوارض دیرینه پیشرفته است. برای تشخیص پدیده Somoga، لازم است سطح گلیسمی حدود 3 ساعت را مطالعه کنید، که جزء جدایی ناپذیر از انتخاب درمان انسولین است. اگر کاهش هیپوگلیسمی شبانه از نظر توسعه هیپوگلیسمی شب همراه باشد، با هیپرگلیسمی در صبح همراه است (پدیده صبح صبح)، بیمار باید یک افزایش زودرس (6-7 صبح) را توصیه کند در حالی که انسولین یک شبه را معرفی کرد، همچنان به حفظ سطح طبیعی گلیسمی ادامه می دهد.
تزریق دوم انسولین NPH معمولا قبل از صبحانه همراه با تزریق صبحانه از انسولین یک اقدام کوتاه (Ultrashort) انجام می شود. در این مورد، دوز به طور عمده بر اساس سطح گلیسمی در مقابل غذاهای اصلی روز (ناهار، شام) انتخاب شده است. علاوه بر این، می تواند توسعه هیپوگلیسمی در فواصل بین وعده های غذایی، به عنوان مثال، در ظهر، بین صبحانه و ناهار، محدود شود.
تمام انسولین دوز اقدام طولانی مدت(Glargin) یک بار در روز معرفی می شود، در حالی که اساسا نه، در چه زمانی. سینتیک
انسولین های گلورژین و دیتک از نظر خطر ابتلا به هیپوگلیسمی، از جمله شب، مطلوب تر هستند.
دوز انسولین کوتاه یا فوق العاده حتی در اولین بیمار، مقصد انسولین به مقدار کربوهیدرات های مورد استفاده (واحد نان) و سطح گلیسمی قبل از تزریق بستگی دارد. به طور شرطی، بر اساس ریتم روزانه ترشح انسولین، حدود 1/4 از انسولین کوتاه مدت (6-8 واحد) به شام \u200b\u200bداده می شود، دوز باقی مانده تقریبا به طور مساوی به صبحانه و ناهار تقسیم می شود (10-12 واحد). بالاتر از سطح اولیه گلیسمی، کمتر آن را توسط واحد انسولین وارد شده کاهش می یابد. تزریق یک اثر کوتاه انسولین 30 دقیقه قبل از غذا، عمل فوق العاده فوق العاده قبل از غذا یا حتی بلافاصله پس از غذا ساخته شده است. کفایت یک دوز انسولین کوتاه مدت از لحاظ گلیسمی 2 ساعت پس از غذا و قبل از غذا بعدی تخمین زده می شود.
برای محاسبه دوز انسولین با درمان شدید انسولین، به اندازه کافی تعداد x را بر اساس جزء کربوهیدرات شمارش می کند. در عین حال، تمام محصولات کربوهیدرات به حساب نمی آیند، اما تنها به اصطلاح محاسبه شده است. دومی شامل سیب زمینی، محصولات دانه، میوه ها، لبنیات مایع و محصولات شیرین است. محصولات حاوی کربوهیدرات های کم ارزش (بیشتر سبزیجات) به حساب نمی آیند. جداول تبادل ویژه توسعه یافته است، با کمک آن، بیان مقدار کربوهیدرات در او می توانید دوز لازم انسولین را محاسبه کنید. یک XE مربوط به 10-12 گرم کربوهیدرات (جدول 10.7) است.
پس از مصرف غذا حاوی 1 X، سطح گلیسمی با 1.6-2.2 mmol / l افزایش می یابد، I.E. تقریبا همانطور که سطح گلوکز کاهش می یابد، زمانی که معرفی 1 U. انسولین کاهش می یابد. به عبارت دیگر، بر روی هر هی، که در غذا قرار دارد، که برنامه ریزی شده برای خوردن است، باید پیش از آن (بسته به زمان روز) حدود 1 واحد انسولین معرفی شود. علاوه بر این، ما باید نتایج خود کنترل سطح گلیسمی را که قبل از هر تزریق انجام می شود، ثبت کنیم و زمان روز (حدود 2 واحد انسولین بر روی 1 HEB در صبح و در ناهار، 1 واحد در 1 X - برای شام). بنابراین، اگر هیپرگلیسمی نشان داده شود، دوز انسولین، محاسبه شده با توجه به وعده غذایی آینده (از لحاظ تعداد XE)، باید افزایش یابد، و بالعکس، اگر هیپوگلیسمی نشان داده شود، انسولین کمتر معرفی می شود.
جدول. 7.7.جایگزینی معادل محصولات تولید کننده 1 x
 به عنوان مثال، اگر یک بیمار 30 دقیقه به شام \u200b\u200bبرنامه ریزی شده حاوی 5 Hehe، سطح گلیسمی 7 میلی مول در لیتر باشد، باید 1 واحد به گلیسمی برای کاهش سطح طبیعی معرفی شود: از 7 mmol / l به حدود 5 mmol / l علاوه بر این، 5 Uzinulin باید در پوشش 5 X معرفی شود. بنابراین، بیمار در این مورد 6 واحد یک عمل کوتاه یا فوق العاده را معرفی می کند.
به عنوان مثال، اگر یک بیمار 30 دقیقه به شام \u200b\u200bبرنامه ریزی شده حاوی 5 Hehe، سطح گلیسمی 7 میلی مول در لیتر باشد، باید 1 واحد به گلیسمی برای کاهش سطح طبیعی معرفی شود: از 7 mmol / l به حدود 5 mmol / l علاوه بر این، 5 Uzinulin باید در پوشش 5 X معرفی شود. بنابراین، بیمار در این مورد 6 واحد یک عمل کوتاه یا فوق العاده را معرفی می کند.
پس از تظاهرات SD-1 و شروع درمان انسولین، به مدت طولانی نیاز به انسولین ممکن است کوچک باشد و کمتر از 0.3-0.4 واحد / کیلوگرم باشد. این دوره به عنوان یک مرحله از بهبودی نشان داده شده است یا "ماه عسل".پس از دوره هیپرگلیسمی و کتواسییدوز، که ترشح انسولین را از بین می برد 10 تا 15 درصد از طریق سلول های حفظ شده بتا، جبران اختلالات فلزات هورمونی با معرفی انسولین، عملکرد این سلول ها را بازسازی می کند، که پس از آن فرضیه یک بدن انسولین را در نظر می گیرد حداقل سطح. این دوره می تواند از چند هفته تا چند سال ادامه یابد، اما در نهایت، به دلیل تخریب اتوایمیون از سلول های باقی مانده بتا، "ماه عسل" به پایان می رسد.
رژیم غذاییبا SD-1 در بیماران آموزش دیده که دارای مهارت های خودکشی و انتخاب دوز انسولین هستند، ممکن است آزادانه باشد، I.E. نزدیک شدن به رایگان اگر بیمار دارای کمبود وزن بیش از حد یا وزن بدن باشد، رژیم غذایی باید باشد
ایزوکلوریایی جزء اصلی مواد غذایی در SD-1 کربوهیدرات است که باید حدود 65 درصد از کالراژ روزانه داشته باشد. اولویت باید به محصولات حاوی کربوهیدرات های پیچیده، به آرامی مکش، و همچنین محصولات غنی از بافت غذایی داده شود. محصولات حاوی کربوهیدرات (آرد، شیرین) باید اجتناب شود. نسبت پروتئین ها باید به 10-35٪ کاهش یابد، که به کاهش خطر ابتلا به میکروآنژیوپاتی و سهم چربی ها تا 25 تا 35 درصد کمک می کند و چربی های محدود باید تا 7 درصد از کالراژ را تشکیل دهند ، که خطر ابتلا به آترواسکلروز را کاهش می دهد. علاوه بر این، لازم است از مصرف نوشیدنی های الکلی، به ویژه قوی، اجتناب شود.
جزء یکپارچه کار با یک بیمار با SD-1 و تعهد جبران مؤثر آن است آموزش بیماراندر طول عمر، بیمار باید روزانه به طور مستقل به عوامل متعددی بستگی دارد تا دوز انسولین را تغییر دهد. بدیهی است، به داشتن مهارت های خاصی که بیمار باید آموزش داده شود، نیاز دارد. "مدرسه SD-1 بیمار" در بیمارستان های غدد درون ریز یا سرپایی سازماندهی شده است و نشان دهنده 5-7 کلاس ساختاری است که در آن پزشک یا یک پرستار مخصوص آموزش دیده در حالت تعاملی با استفاده از مزایای مختلف بصری، اصول آموزش بیمار را انجام می دهد selfOntrol
پیش بینی
در غیاب درمان انسولین، بیمار SD-1 به ناچار از کما کتواسیدیوتیک میمیرد. با درمان ناکافی انسولین، در برابر پس زمینه که معیارهای جبران CD به دست نمی آید و بیمار در حالت هیپرگلیسمی مزمن قرار دارد (جدول 7.3)، عوارض دیررس (§ 7.8) شروع به توسعه و پیشرفت می کند. با SD-1، بیشترین اهمیت بالینی در این زمینه، تظاهرات میکرودیسم دیابتی (نفروپاتی و رتینوپاتی) و نوروپاتی (سندرم پا دیابتی) را نشان می دهد. Macroangiopathy با SD-1 در پیش زمینه نسبتا نادر است.
7.6. دیابت نوع 2
دیابت نوع 2- یک بیماری مزمن نشان دهنده نقض مبادلات کربوهیدرات با توسعه هیپرگلیسمی به علت مقاومت به انسولین و اختلال عملکرد سلول های β-
و همچنین متابولیسم لیپید با توسعه آترواسکلروز. از آنجا که علت اصلی مرگ و ناتوانی بیماران عوارض آترواسکلروز سیستمیک است، SD-2 گاهی اوقات به عنوان یک بیماری قلبی عروقی نامیده می شود.
جدول. 7.8.دیابت نوع 2
 اتیولوژی
اتیولوژی
SD-2 یک بیماری چند منظوره با استعداد ارثی است. همبستگی در SD-2 در دوقلوهای تک بار 80٪ یا بیشتر می رسد. اکثر بیماران مبتلا به SD-2 نشان دهنده حضور SD-2 برای نزدیکترین بستگان هستند؛ در حضور SD-2 در یکی از والدین، احتمال بروز آن در طول زندگی، 40٪ است. کدام یک ژن، پلی مورفیسم که مستعد ابتلا به SD-2 را تعیین می کند، شناسایی نشده است. عوامل محیطی اهمیت زیادی در اجرای مستعد ابتلا به SD-2 دارد. عوامل خطر ابتلا به SD-2 عبارتند از:
چاقی، به ویژه احشایی (پاراگراف 11.2 را ببینید)؛
قومیت (به ویژه در هنگام تغییر سبک زندگی سنتی در غرب)؛
شیوه زندگی کم تحرک؛
ویژگی های رژیم غذایی (مصرف زیاد کربوهیدرات های تصفیه شده و محتوای فیبر کم)؛
فشار خون شریانی.
پاتوژنز
Pathogenetically SD-2 یک گروه ناهمگن اختلالات متابولیک است، دقیقا مشخص است که ناهمگونی بالینی قابل توجه آن را تعیین می کند. اساس پاتوژنز آن، مقاومت به انسولین (کاهش انسولین متخلخل از استفاده از گلوکز توسط بافت ها) است که در برابر پس زمینه اختلال ترشح سلول های بتا، اجرا می شود. بنابراین، نقض تعادل حساسیت به انسولین و ترشح انسولین وجود دارد. اختلال عملکرد ترشحβ - ناوگاناین است که کاهش انتشار "زودهنگام" انسولین در پاسخ به افزایش گلوکز خون. در همان زمان، مرحله 1 (سریع) ترشح، که در تخلیه حوضچه ها با انسولین انباشته قرار دارد، در واقع غایب است؛ فاز ترشح دوم (آهسته) در پاسخ به تثبیت هیپرگلیسمی به طور مداوم، در حالت تونیک انجام می شود و علیرغم ترشح بیش از حد انسولین، سطح گلیسمی در برابر پس زمینه مقاومت به انسولین طبیعی نیست (شکل 7.8).
نتیجه هیپینسلینمی این است که حساسیت و تعداد گیرنده های انسولین و سرکوب را کاهش دهد
مکانیسم های پس از گیرنده تشویق اثرات انسولین (مقاومت به انسولین).محتوای اصلی نوار نقاله گلوکز در سلول های عضلانی و چربی (GLUT-4) 40٪ در افراد مبتلا به چاقی احشایی و 80٪ در افراد مبتلا به SD-2 کاهش می یابد. با توجه به مقاومت به انسولین hepatocytes و hyperinsulinemia پورتال، گلوکز بیش از حد تولید، کبد،و هیپرگلیسمی در حال توسعه است، که در اکثر بیماران مبتلا به SD-2، از جمله در مراحل اولیه بیماری تشخیص داده می شود.
هیپرگلیسمی خود را به طور قابل توجهی بر ماهیت و سطح فعالیت ترشحی سلول های بتا (گلوکوزوتوکسیسیت) تاثیر می گذارد. برای مدت طولانی، طی سالها و دهه ها، هیپرگلیسمی موجود در نهایت منجر به تخریب سلول های β- سلول های انسولین می شود و برخی از علائم ممکن است در بیمار ظاهر شود. کمبود انسولین- لاغری، کتوز با بیماری های عفونی مرتبط. با این حال، محصولات انسولین باقی مانده، که برای جلوگیری از کتواسیدوز کافی است، تقریبا همیشه در SD-2 حفظ می شود.
همهگیرشناسی
SD-2 اپیدمیولوژی SD را به طور کلی تعریف می کند، زیرا حدود 98 درصد موارد این بیماری را تشکیل می دهد. شیوع SD-2 در کشورهای مختلف و گروه های قومی متفاوت است. در اروپا
 شکل. 7.8.اختلال عملکرد ترشح سلول های بتا با دیابت نوع 2 (از دست دادن مرحله ترشح سریع انسولین 1)
شکل. 7.8.اختلال عملکرد ترشح سلول های بتا با دیابت نوع 2 (از دست دادن مرحله ترشح سریع انسولین 1)
کشورها، ایالات متحده آمریکا و فدراسیون روسیه، حدود 5-6 درصد از جمعیت است. با سن، بروز SD-2 افزایش می یابد: در میان بزرگسالان شیوع SD-2 10٪ است، در میان افرادی که بیش از 65 سال به 20٪ می رسد. بروز SD-2 2.5 برابر بیشتر از افراد بومی امریکا و جزایر هاوایی است. در میان سرخپوستان قبیله پیما (آریزونا) آن را به 50٪ می رسد. در میان جمعیت روستایی هند، چین، شیلی و کشورهای آفریقایی که شیوه زندگی سنتی را رهبری می کنند، شیوع SD-2 بسیار کم است (کمتر از 1٪). از سوی دیگر، در میان مهاجران در کشورهای صنعتی غربی، سطح قابل توجهی را به دست می آورد. بنابراین، در میان مهاجران هند و چین، زندگی در ایالات متحده و انگلستان، شیوع SD-2 به 12-15٪ می رسد.
چه کسی پیش بینی می کند که تعداد بیماران دیابت در جهان در 20 سال آینده 122 درصد افزایش یابد (از 135 تا 300 میلیون نفر). این به دلیل پیری پیشرفته جمعیت و با توزیع و تشدید شیوه زندگی شهری است. در سال های اخیر، "جوان سازی" SD-2 و رشد میزان بروز آن در میان کودکان وجود داشته است.
تظاهرات بالینی
در بیشتر موارد، تظاهرات بالینی تلفظ شده وجود نداردو تشخیص در طول تعریف معمول سطح گلیسمی ایجاد می شود. این بیماری معمولا در سن 40 سالگی ظاهر می شود، در حالی که اکثریت قریب به اتفاق بیماران دارای چاقی و سایر اجزای سندرم متابولیک هستند (نگاه کنید به بند 11.2). بیماران شکایات مربوط به ظرفیت کاری را تحمیل نمی کنند، اگر دلایل دیگری برای این وجود نداشته باشند. شکایات تشنگی و پلیوریا به ندرت به شدت قابل توجه می رسند. اغلب بیماران در مورد پوست و خارش واژن نگران هستند و به همین دلیل آنها به متخصصین پوست و متخصصین پزشکان تبدیل می شوند. از آنجاییکه بسیاری از سالها (به طور متوسط \u200b\u200bحدود 7 سال)، بسیاری از بیماران در زمان تشخیص بیماری در تصویر بالینی، در زمان شناسایی بیماری در تصویر بالینی، اغلب تحت سلطه از تظاهرات واقعی از آن قرار می گیرند SD-2 قبل از تشخیص. علائم و تظاهرات عوارض دیررس SD.علاوه بر این، اولین درخواست تجدید نظر بیمار مبتلا به SD-2 برای کمک های پزشکی اغلب به علت عوارض دیررس است. بنابراین، بیماران می توانند در بیمارستان های جراحی با ضایعات پپتیک پاها بستری شوند (سندرم پا دیابتی)،تماس با پیوند چشم انداز پیشرفته به چشم پزشکان (رتینوپاتی دیابتی)،بستری شده با حملات قلبی، سکته مغزی
تامی، شکست رگ های پا در موسسه، جایی که آنها ابتدا هیپرگلیسمی را پیدا می کنند.
تشخیصی
معیارهای تشخیصی، یکنواخت برای انواع SD، در بند 7.3 ارائه شده است. تشخیص SD-2 در اکثریت قریب به اتفاق بر اساس تشخیص هیپرگلیسمی در افراد مبتلا به علائم بالینی معمول SD-2 (چاقی، سن بیش از 40-45 سال، سابقه خانوادگی مثبت SD-2، سایر اجزای سندرم متابولیک است )، در غیاب علائم بالینی و آزمایشگاهی کمبود مطلق انسولین (کاهش وزن، کتوز) را نشان می دهد. ترکیبی از شیوع بالای SD-2، مشخصه یک جریان بدون علامت طولانی و امکان جلوگیری از عوارض سنگین او، با توجه به تشخیص زودهنگام، از پیش تعیین شده نیاز دارد غربالگریکسانی که. معاینه به منظور حذف SD-2 در میان افراد بدون علائم بیماری. آزمون اصلی، به عنوان نشان داده شده، تعریف است سطح گلیسمی معده خالی است.این در شرایط زیر نشان داده شده است:
1. همه افراد بالای 45 سال سن دارند، به خصوص بیش از وزن بدن (CMT بیش از 25 کیلوگرم در متر مربع) در فواصل هر 3 سال.
2. در کوچکترین سن در حضور وزن بیش از حد بدن (BMI با بیش از 25 کیلوگرم در متر مربع) و عوامل خطر اضافی که شامل موارد زیر می شود
شیوه زندگی کم تحرک؛
SD-2 برای نزدیکترین بستگان؛
متعلق به خطر ملی توسعه SD-2 (آمریکایی های آفریقایی آمریکایی، آمریکای لاتین، آمریکایی های بومی و غیره)؛
زنان که یک کودک را با وزن بیش از 4 کیلوگرم و / یا در حضور دیابت حاملگی به عنوان یک تاریخ به دنیا آوردند؛
فشار خون شریانی (≥ 140/90 میلیمتر Hg)؛
سطح HDL\u003e 0.9 mmol / L و / یا تری گلیسیرید\u003e 2.8 mmol / l؛
سندرم تخمدان پلی کیستیک؛
NTG و NGN؛
بیماری های قلبی عروقی.
افزایش قابل توجهی در بروز SD-2 در میان کودکان، نیاز به تعریف غربالگری سطح گلیسمی را تعیین می کند در میان کودکان و نوجوانان(از 10 سال با فاصله 2 سال یا با شروع
pubertata، اگر او در سن زودرس رخ داد) متعلق به گروه های افزایش خطر ابتلا به کودکان است با فراوانی وزن بدن(BMI و / یا وزن بدن\u003e 85 درصد، سن مناسب یا وزن بیش از 120 درصد نسبت به ایده آل) در ترکیب با هر دو عامل خطر اضافی ذکر شده است:
SD-2 در میان بستگان خط اول یا دوم خویشاوندی؛
متعلق به ملیت های پر خطر؛
تظاهرات بالینی مرتبط با مقاومت به انسولین (Acanthosis Nigricans،پرفشاری خون شریانی، دیس لیپیدمی)؛
SD، از جمله حاملگی، مادر.
تشخیص های افتراقی
تشخیص افتراقی SD-2 و SD-1، اصول آن در بند 7.5 شرح داده شده در بند 7.5 (جدول 7.6) شرح داده شده است. همانطور که نشان داده شده است، در اغلب موارد بر اساس داده های تصویر بالینی است. در مواردی که استقرار نوع سی دی با مشکلات مواجه می شود، یا سوء ظن برخی از انواع نادر SD، از جمله در چارچوب سندرم های ارثی، مهمترین مسئله عملی است که لازم است پاسخ آن این است که آیا بیمار است نیاز به یک بیمار در درمان انسولین دارد.
رفتار
مولفه های اصلی درمان SD-2 عبارتند از: رژیم غذایی درمانی، گسترش فعالیت بدنی، درمان قند، پیشگیری و درمان عوارض دی اکسید کربن. از آنجایی که اکثر بیماران مبتلا به SD-2 از چاقی رنج می برند، رژیم غذایی باید با هدف کاهش وزن (هیپوچالر) و پیشگیری از عوارض دیررس، عمدتا Macroangiopathy (آترواسکلروز) باشد. رژیم غذایی هیپولیزبرای همه بیماران با وزن بیش از حد بدن (BMI 25-29 کیلوگرم در متر مربع) یا چاقی (BMI\u003e 30 کیلوگرم در متر مربع) ضروری است. در بیشتر موارد، باید توصیه شود تا لبه های روزانه غذا را به 1000-1200 کیلوکالری برای زنان و تا 1200-1600 کیلوکالری برای مردان کاهش دهد. نسبت توصیه شده اجزای اصلی مواد غذایی در SD-2 مشابه آن با SD-1 (کربوهیدرات - 65٪، پروتئین 10-35٪، چربی تا 25-35٪) است. استفاده کنید الکللازم است که به علت این واقعیت محدود شود که منبع ضروری کالری های اضافی است، علاوه بر این، پذیرش الکل در پس زمینه TERA
fDI با سولفونیل اوره و انسولین می تواند توسعه هیپوگلیسمی را تحریک کند (به پاراگراف 7.7.3 مراجعه کنید).
توصیه برای گسترش فعالیت بدنیباید فردی باشد در ابتدا، بارهای هوازی (پیاده روی، شنا، شنا) مدت زمان متوسط \u200b\u200b30-45 دقیقه 3-5 بار در روز (حدود 150 دقیقه در هفته) توصیه می شود. در آینده، ضروری است که به تدریج اعمال فیزیکی را افزایش دهیم، که به طور قابل توجهی به کاهش وزن بدن کمک می کند. علاوه بر این، اعمال فیزیکی به کاهش مقاومت به انسولین کمک می کند و اثر هیپوگلیسیم را کاهش می دهد. ترکیبی از رژیم غذایی و گسترش اعمال فیزیکی بدون انتصاب داروهای قند، امکان حفظ جبران SD را مطابق با اهداف ایجاد شده (جدول 7.3) تقریبا 5٪ از بیماران مبتلا به SD-2 فراهم می کند.
آماده سازی برای درمانی سنیهنگامی که SD-2 را می توان به چهار گروه اصلی تقسیم کرد.
I. آماده سازی کمک به کاهش مقاومت به انسولین (حساسیت).این گروه شامل متفورمین و تیاازولیدیون ها است. متفورمینتنها کسی است که در حال حاضر توسط دارو از گروه استفاده می شود biguanids.اجزای اصلی مکانیسم عمل آن عبارتند از:
1. سرکوب گلوکونوژنز در کبد (کاهش محصولات گلوکز با کبد)، که منجر به کاهش سطح گلیسمی بر روی معده خالی می شود.
2. کاهش مقاومت به انسولین (افزایش دفع گلوکز توسط بافت های محیطی، عمدتا با عضلات).
3. فعال سازی گلیکولیز Avnaeobic و کاهش مکش گلوکز در روده کوچک.
متفورمیناین آماده سازی اولین انتخاب شکر در بیماران مبتلا به SD-2، چاقی و هیپرگلیسمی بر روی معده خالی است. دوز اولیه 500 میلی گرم در هر شب یا در طول شام است. در آینده، دوز به تدریج به 2-3 گرم برای 2-3 پذیرش افزایش می یابد. در میان عوارض جانبی نسبتا اغلب پدیده های دیسپپتیک (اسهال) وجود دارد که به عنوان یک قاعده، به طور مستقل و به طور مستقل پس از 1-2 هفته دریافت دارو دریافت می شود. از آنجایی که متفورمین اثر تحریک کننده ای بر محصولات انسولین ندارد، در پس زمینه مونوتراپی با استفاده از این هیپوگلیسمی دارو
توسعه (عمل آن به عنوان antihyperglycemic تعیین شده است، و نه به عنوان هیپوگلیسمی). منع مصرف متفورمین، حاملگی، قلب شدید، کبد، کلیوی و سایر نارسایی های ارگان، و همچنین حالت های هیپوکسیک یکی دیگر از پیدایش ها هستند. یک عارضه بسیار نادر، که زمانی رخ می دهد که متفورمین بدون در نظر گرفتن منع مصرف های ارائه شده، لاکتاتاکایدوز است، که نتیجه آن یک نتیجه از هیپرووراسیون گلیکولیز بی هوازی است.
thiazolidindions(Pioglitazone، Rosigtyazon) آگونیست های گیرنده های γ فعال شده توسط پراکسیز (PPAR-γ) هستند. Thiazolidindions متابولیسم گلوکز و لیپیدها را در بافت های عضلانی و چربی فعال می کند که منجر به افزایش فعالیت انسولین اندوژن می شود، I.E. برای از بین بردن مقاومت به انسولین (حساسیت های انسولین). دوز روزانه pioglitazone 15-30 میلی گرم در روز، roseglitazone - 4-8 میلی گرم (در هر 1-2 پذیرش) است. ترکیبی از تیاازولیدین ها با متفورمین بسیار موثر است. منع مصرف THIAZOLIDINGION افزایش (2.5 برابر یا بیشتر) سطح ترانس آمیناز های کبدی است. علاوه بر سمیت کبد، عوارض جانبی تیاازولیدین شامل تاخیر در مایع و تورم است که اغلب در طول ترکیبی از آماده سازی انسولین رشد می کنند.
دوم آماده سازی موثرβ - بهتر و کمک به تقویت ترشح انسولین.این گروه شامل آماده سازی سولفونیل اوره و رس (رگولاتورهای گلیقیم پرولامی) است که عمدتا به منظور عادی سازی سطح گلیسمی پس از غذا استفاده می شود. هدف اصلی آماده سازی سولفونیل موچین ها(PSM) β- سلول های جزایر پانکراس هستند. PSM به غشای بتا سلول با گیرنده های خاص متصل می شود. این منجر به بسته شدن کانال های پتاسیم وابسته به ATP و depolarization غشای سلولی می شود که به نوبه خود به باز شدن کانال های کلسیم کمک می کند. جریان کلسیم داخل سلول های بتا منجر به تخریب انسولین و انتشار انسولین به خون می شود. در عمل بالینی، بسیار زیاد از PSMS استفاده می شود، که در طول مدت و شدت اثر قند متفاوت است (جدول 7.9).
جدول. 7.9.آماده سازی سولفونیل موچین ها
 عوارض جانبی اصلی و نسبتا مکرر PSM هیپوگلیسمی است (به بند 7.7.3 مراجعه کنید). این می تواند با مصرف بیش از حد دارو، تجمع آن (نارسایی کلیه) رخ دهد
عوارض جانبی اصلی و نسبتا مکرر PSM هیپوگلیسمی است (به بند 7.7.3 مراجعه کنید). این می تواند با مصرف بیش از حد دارو، تجمع آن (نارسایی کلیه) رخ دهد
عدم انطباق با رژیم غذایی (گذراندن غذا، مصرف الکل) یا حالت (اعمال فیزیکی قابل توجه، قبل از آن دوز PSM کاهش نمی یابد یا کربوهیدرات ها گرفته نمی شود).
به گروه هینیدا(تنظیم کننده های پیشگیرانه گلیسمی) repaglinide(مشتق اسید بنزوئیک؛ دوز روزانه 0.5-16 میلی گرم در روز) و نایفیلینیدا(D-phenylalanine مشتق شده؛ دوز روزانه 180-540 میلی گرم در روز). پس از مصرف مواد مخدر، آماده سازی به سرعت و برگشت پذیر با گیرنده سولفونیلیرولین بر روی سلول β، به عنوان یک نتیجه افزایش کوتاه در سطح انسولین رخ می دهد، که تقلید مرحله اول ترشح آن طبیعی است. آماده سازی در 10-20 دقیقه به وعده های غذایی اصلی، معمولا 3 بار در روز پذیرفته می شود.
III آماده سازی که باعث کاهش جذب گلوکز در روده می شود.
این گروه شامل رزین Akaboz و Guar است. مکانیسم عمل Acarbosis، محاصره برگشت پذیر از α-glycosidases از روده کوچک است، در نتیجه فرآیند تخمیر و مکش های کربوهیدرات متوالی کاهش می یابد، سرعت جذب و پذیرش گلوکز به کبد کاهش می یابد و سطح گلیسمی پس از تزریق کاهش می یابد. دوز اولیه Acarbosis 50 میلی گرم 3 بار در روز، در آینده، دوز می تواند به 100 میلی گرم 3 بار در روز افزایش یابد؛ این دارو بلافاصله قبل از خوردن غذا یا در طول غذا پذیرفته می شود. اثر جانبی اصلی Acarbosa، دیسپپسی روده ای است که با دریافت کربوهیدرات های غیر کشف شده به روده بزرگ همراه است. اثر پارامتری آکربوز بسیار متوسط \u200b\u200bاست (جدول 7.10).
در عمل بالینی، داروهای قابل انعطاف تبلت به طور موثری با یکدیگر و با آماده سازی انسولین ترکیب می شوند، زیرا اکثر بیماران به طور همزمان به عنوان هیپرگلیسمی مكمل و پس از زایمان تعریف می شوند. تعداد زیادی وجود دارد ترکیبات ثابتآماده سازی در یک قرص. اغلب در یک قرص، متفورمین با PSMS های مختلف، و همچنین متفورمین با تیاازولیدین ها ترکیب شده است.
جدول. 7.10.مکانیسم عمل و کارایی بالقوه داروهای شکر قرص
 IV انسولین ها و آنالوگ های انسولین
IV انسولین ها و آنالوگ های انسولین
در مرحله خاصی، آماده سازی انسولین شروع به دریافت تا 30-40٪ بیماران مبتلا به SD-2 می شود. علائم درمان انسولین در SD-2 در ابتدای پاراگراف 7.4 ارائه شده است. رایج ترین گزینه برای ترجمه بیماران مبتلا به SD-2 در درمان انسولین، تعیین انسولین عمل طولانی مدت (NPH انسولین، گلورژین یا دتوکلاسیون) در ترکیب با داروهای مبتنی بر شکر دریافت شده است. در شرایطی که سطح گلیسمی امکان کنترل انتصاب متفورمین یا آخرین ممنوعیت را کنترل نمی کند، بیمار به تزریق انسولین شبانه (یک شبه) اختصاص داده می شود. اگر با کمک داروهای تبلتیک به عنوان یک جیوه و گلیسمی پس از تزریق غیرممکن باشد، بیمار به یک مونوسلیناتراپی ترجمه می شود. معمولا با درمان انسولین SD-2 بر روی آن به اصطلاح انجام می شود طرح "سنتی"که به معنای انتصاب دوزهای ثابت از انسولین طولانی مدت و کوتاه است. در این طرح
مخلوط های استاندارد انسولین حاوی یک عمل کوتاه (UltraShort) و طولانی مدت در یک بطری مناسب هستند. انتخاب درمان انسولین سنتی توسط این واقعیت که تحت SD-2 تعیین می شود، اغلب به بیماران سالمند منصوب می شود که یادگیری به یک تغییر مستقل در دوز انسولین دشوار است. علاوه بر این، درمان انسولین شدید، هدف آن حفظ جبران تبادل کربوهیدرات در سطح نزدیک شدن به نوروگلیسمی، خطر ابتلا به هیپوگلیسمی را افزایش می دهد. اگر برای بیماران جوان، هیپوگلیسمی نور، خطر جدی را نشان نمی دهد، در بیماران سالمند مبتلا به آستانه کاهش حساسیت هیپوگلیسمی، آنها می توانند اثرات بسیار نامطلوب از سیستم قلبی عروقی داشته باشند. بیماران جوان مبتلا به SD-2، و همچنین بیماران از لحاظ فرصت های یادگیری موثر، نسخه فشرده درمان انسولین را می توان منصوب کرد.
پیش بینی
دلیل اصلی معلولیت و مرگ بیماران مبتلا به SD-2 عوارض دیررس است (به بند 7.8 مراجعه کنید)، اغلب Macroangiopathy دیابتی. خطر ابتلا به برخی از عوارض بعد از آن، توسط پیچیده ای از عوامل مورد بحث در فصل های مربوطه تعیین می شود. یک عامل خطر جهانی برای توسعه آنها هیپرگلیسمی مزمن است. بنابراین، کاهش سطح HbA1c در بیماران مبتلا به SD-2 در هر 1٪ منجر به کاهش کل مرگ و میر در حدود 20٪، به ترتیب 2٪ و 3٪ - به ترتیب 40٪
7.7. عوارض حاد دیابت
7.7.1. کتواسییدوز دیابتی
کتواسییدوز دیابتی (DCA)- تخفیف SD-1، به علت کمبود مطلق انسولین، در غیاب درمان به موقع کم کاری کتوسیدیوتیک (QC) و مرگ.
اتیولوژی
علت DCA کمبود انسولین مطلق است. این یا شدت DCA در اکثر بیماران در زمان تظاهرات SD-1 (10-20٪ موارد DCA) تعیین می شود.
بیمار مبتلا به تشخیص تشخیص DCA SD-1 می تواند زمانی که انسولین متوقف شود، اغلب توسط بیمار خود (13٪ موارد DCA)، در پس زمینه بیماری های همزمان، عمدتا عفونی، در صورت عدم افزایش انسولین، متوقف می شود دوز
جدول. 7.11.کتواسییدوز دیابتی
 تا 20 درصد از موارد توسعه DC-1 در بیماران جوان مبتلا به SD-1 با مشکلات روانشناختی و / یا اختلالات رفتار غذا (ترس از افزایش وزن، ترس از هیپوگلیسمی، مشکلات نوجوان) همراه است. علت مکرر DCA در تعدادی از کشورها است
تا 20 درصد از موارد توسعه DC-1 در بیماران جوان مبتلا به SD-1 با مشکلات روانشناختی و / یا اختلالات رفتار غذا (ترس از افزایش وزن، ترس از هیپوگلیسمی، مشکلات نوجوان) همراه است. علت مکرر DCA در تعدادی از کشورها است
لغو انسولین توسط بیمار خود به دلیل هزینه بالای مواد مخدر برای برخی از بخش های جمعیت (جدول 7.11).
پاتوژنز
در قلب پاتوژنز DCA، کمبود مطلق انسولین را در ترکیب با افزایش محصولات هورمونهای کنونی مانند گلوکاگون، کاتچولامین و کورتیزول قرار می دهد. در نتیجه، افزایش قابل توجهی در محصولات کبد گلوکز و اختلال دفع آن با بافت های محیطی، افزایش هیپرگلیسمی و اختلال اسمولاریسی فضای خارج سلولی وجود دارد. کمبود انسولین در ترکیب با بیش از حد نسبی از هورمون های متضاد در DCA منجر به انتشار اسید های چرب آزاد (لیپولیز) و اکسیداسیون غیر فشرده سازی آنها در کبد به بدن کتون (β- هیدروکسی بوتیرات، استئوآساتات، استون)، به عنوان یک نتیجه از آن hypercohememia توسعه می یابد، و در آینده اسیدوز متابولیک. به عنوان یک نتیجه از غلظت گلوکز، diuresis اسمزی، deursdration، از دست دادن سدیم، پتاسیم و سایر الکترولیت ها (شکل 7.9) توسعه می یابد.
همهگیرشناسی
فراوانی موارد جدید DCA 5-8 در هر 1000 بیمار مبتلا به SD-1 در سال است و به طور مستقیم بستگی به سطح سازمان بیماران مراقبت های پزشکی با SD دارد. هر سال در ایالات متحده حدود 100،000 بیمارستان بستری در مورد DCA رخ می دهد، در حالی که با توجه به هزینه های یک بیمار برای بستری شدن 13 هزار دلار، بیش از 1 میلیارد دلار در سال سالانه در درمان ثابت DCA صرف می شود. در فدراسیون روسیه در سال 2005، DKA در 4.31٪ از کودکان، 4.75٪ از نوجوانان و 0.33٪ از بیماران بالغ با SD-1 ثبت شد.
تظاهرات بالینی
بسته به علت علت، بسته به علت ناشی از دلیل آن، می تواند از چند هفته تا روز باشد. در اغلب موارد، DCA پیش از علائم دیابت است، اما گاهی اوقات ممکن است وقت خود را برای توسعه نداشته باشند. علائم بالینی DCA شامل پلیوریا، پلیدیپسی، لاغری، درد شکمی ریخته شده ("pseudoperithonite دیابتی")، کم آبی، ضعف ضخیم، بوی استون از دهان (یا بوی میوه)، به تدریج آگاهی را به دست می آورند. کاما واقعی در DCA به تازگی به دلیل تشخیص زودهنگام نسبتا به ندرت توسعه یافته است. در تحقیقات فیزیکی، نشانه های کم آبی بدن شناسایی می شود: کاهش
 شکل. 7.9. پاتوژنز کما کتوسیدیوتیک
شکل. 7.9. پاتوژنز کما کتوسیدیوتیک
turgor از پوست و تراکم چشم، تاکیکاردی، هیپوتانسیون. در موارد در حال ظهور، تنفس Kussmouul توسعه می یابد. بیش از 25٪ از بیماران مبتلا به DCA استفراغ را توسعه می دهند، که می تواند شبیه قهوه ضخیم در رنگ باشد.
تشخیصی
این بر اساس داده های تصویر بالینی، نشانه های حضور SD-1 بیمار، و همچنین داده های مطالعه آزمایشگاهی است. برای DCA، hyperglycemia مشخص می شود (در برخی موارد، ناچیز)، کتونوری، اسیدوز متابولیک، هیپروسومولاری (جدول 7.12).
جدول. 7.12.تشخیص آزمایشگاهی عوارض شدید دیابت
 هنگام بررسی بیماران مبتلا به کمبود شدید SD، تعیین سطح گلیسمی، کراتینین و اوره، الکترولیت ها، بر اساس آن اسمولاریسی موثر محاسبه می شود. علاوه بر این، یک اسکورت و حالت زمین مورد نیاز است. اسمولاریس موثر(EO) بر اساس فرمول زیر محاسبه می شود: 2 *. Norma Eo 285 - 295 MOSM / L است.
هنگام بررسی بیماران مبتلا به کمبود شدید SD، تعیین سطح گلیسمی، کراتینین و اوره، الکترولیت ها، بر اساس آن اسمولاریسی موثر محاسبه می شود. علاوه بر این، یک اسکورت و حالت زمین مورد نیاز است. اسمولاریس موثر(EO) بر اساس فرمول زیر محاسبه می شود: 2 *. Norma Eo 285 - 295 MOSM / L است.
اکثر بیماران مبتلا به DCA تعیین می شوند لکوسیتوزشدت آن متناسب با سطح بدن کتون در خون است. مرحله سدیمبه عنوان یک قاعده، به دلیل خروج مایع اسموتی از فضاهای داخل سلولی به خارج از سلولی در پاسخ به هیپرگلیسمی کاهش می یابد. کمتر از سطح سدیم می تواند به عنوان یک نتیجه از بیش از حد تلفظ شده به اشتباه کاهش یابد
تری گلیسیریدمی مرحله پتاسیمسرم می تواند در ابتدا به دلیل حرکت آن از فضاهای خارج سلولی افزایش یابد.
تشخیص های افتراقی
دلایل دیگر از دست دادن آگاهی در بیماران مبتلا به SD. تشخیص دیفرانسیل با کما هیپرونسمولار، به عنوان یک قاعده، باعث مشکلات نمی شود (در بیماران سالمند مبتلا به SD-2 توسعه می یابد) و ارزش کلینیکی بزرگ را ندارد، زیرا اصول درمان هر دو حالت مشابه هستند. اگر غیرممکن باشد که به سرعت متوجه شود دلیل از دست دادن آگاهی بیمار با SD، معرفی گلوکز از آن زمان نشان داده شده است ایالت های هیپوگلیسمی خیلی بیشتر یافت می شوند و یک پویایی مثبت سریع در برابر پس زمینه معرفی گلوکز به ما اجازه می دهد تا دلیل از دست دادن آگاهی را بیابیم.
رفتار
درمان DKA نشان می دهد که Rehydration، اصلاح هیپرگلیسمی، اختلالات الکترولیت، و همچنین درمان بیماری هایی که باعث کاهش میزان دیابت می شود. درمان بیشتر به طور مطلوب در بخش احیاء موسسه پزشکی تخصصی انجام می شود. در بیماران بالغ بدون آسیب شناسی قلب شدید، هنوز هم در مرحله قبل از بیمارستان به عنوان یک اقدام اولیه برای هدف ریهتوصیه می شود یک راه حل ایزوتونیک (0.9٪ NaCl) را تقریبا با سرعت یک لیتر در ساعت (حدود 15-20 میلی لیتر در هر کیلوگرم وزن در ساعت) معرفی کنید. بازپرداخت کامل کمبود مایع، که با DCA 100-200 میلی لیتر در کیلوگرم وزن است، باید در روزهای اول درمان به دست آید. با نارسایی همزمان قلب یا کلیه، این دوره زمان باید افزایش یابد. برای کودکان، حجم توصیه شده از راه حل ایزوتونیک برای درمان مجدد درمانی 10 تا 20 میلی لیتر در هر کیلوگرم وزن بدن در ساعت است و در 4 ساعت اول نباید بیش از 50 میلی لیتر در کیلوگرم وزن باشد. Rehydration کامل توصیه می شود برای رسیدن به حدود 48 ساعت. پس از آنکه در پس زمینه درمان انسولین موازی، سطح گلیسمی در حدود 14 میلی مول در لیتر کاهش می یابد، به انتقال یک محلول گلوکز 10٪، که همچنان به احیای مجدد ادامه می یابد، کاهش می یابد.
در حال حاضر مفهوم "دوزهای کوچک" را پذیرفته است انسولیندر درمان DCA. فقط یک انسولین کوتاه عمل استفاده می شود. بهینه ترین استفاده از مدارک داخل وریدی
لینا معرفی داخل عضلانی انسولین، که کمتر کارآمد است، احتمالا تنها با شدت متوسط \u200b\u200bDCA، با همودینامیک پایدار و اگر غیر ممکن برای درمان داخل وریدی باشد. در مورد دوم، تزریق در عضله مستقیم شکم ساخته می شود، در حالی که سوزن تزریق عضلانی بر روی یک سرنگ انسولین قرار می گیرد (برای یک ضربه عضلانی قابل اعتماد) و در این سوزن، انسولین از بطری به سرنگ استخدام می شود .
چندین گزینه برای تزریق انسولین داخل وریدی وجود دارد. اول، انسولین را می توان "به گروه لاستیکی" سیستم تزریق معرفی کرد، در حالی که مقدار مورد نیاز انسولین به یک سرنگ انسولین به دست می آید، پس از آن 1 میلی لیتر از راه حل ایزوتونیک به دست می آید. تا رسیدن به سطح گلیسمی، 14 mmol / l یک بیمار ساعتی است که 6 تا 10 واحد از اقدام کوتاه معرفی شده است؛ به علاوه (به موازات تغییر محلول Rehydration با ایزوتونیک با 10٪ گلوکز)بسته به زمان شاخص های قابل تعریف گلیسمی، دوز انسولین به 4-8 واحد در ساعت کاهش می یابد. میزان کاهش توصیه شده سطح گلیسمی نباید بیش از 5 mmol / l در ساعت باشد. یکی دیگر از تجسم درمان انسولین داخل وریدی، استفاده از Perfuzor را نشان می دهد. برای آماده سازی permetator، 2 میلی لیتر از محلول 20٪ از راه حل آلبومین انسان به 50 میلی گرم محلول ایزوتونیک 0.9٪ اضافه می شود. اگر مسیر عضلانی تزریق انسولین انتخاب شود، 20 واحد یک عمل کوتاه ابتدا در ابتدا معرفی می شود، پس از آن 6 واحد، و پس از رسیدن به سطح گلیسمی، 14 میلی مول در لیتر دوز به 4 واحد در ساعت کاهش می یابد. پس از تثبیت کامل همودینامیک و جبران خسارت اختلالات اسید-پایه، بیمار به تزریق انسولین زیر جلدی ترجمه می شود.
همانطور که نشان داده شده است، علیرغم قابل توجه کمبود پتاسیمدر بدن (از دست دادن کلی 3-6 میلی مول / کیلوگرم)، با سطح DCA خود را قبل از شروع درمان انسولین می تواند تا حدودی افزایش یابد. با این وجود، شروع انتقال خون محلول کلرید پتاسیم به طور همزمان با شروع درمان انسولین انجام می شود، اگر سطح پتاسیم پلاسما کمتر از 5.5 mmol / l باشد. اصلاح موفقیت آمیز کمبود پتاسیم تنها در برابر پس زمینه نرمال سازی pH رخ می دهد. با استفاده از pH پایین، مصرف پتاسیم به طور قابل توجهی کاهش می یابد، در صورت امکان، در صورت امکان، دوز کلرید پتاسیم سرریز مطلوب است که به یک شاخص PH خاص سازگار باشد (جدول 7.13).
جدول. 7.13.طرح تصحیح کمبود پتاسیم
 * برای محاسبه، داده های زیر استفاده می شود:
* برای محاسبه، داده های زیر استفاده می شود:
1 g kcl \u003d 13.4 mmol؛ 1 mmol kcl \u003d 0.075 گرم. در یک محلول 4٪ KS1: در 100 میلی لیتر - 4 گرم Ks1، در 25 میلی لیتر - 1 گرم Ks1، در 10 میلی لیتر از 0.4 گرم Ks1.
دلیل عدم توانایی دیابت اغلب است بیماری های عفونی(پیلونفریت، زخم آلوده با سندرم پا دیابتی، پنومونی، سینوزیت و غیره). یک قاعده وجود دارد که با آن، با آنتی بیوتیک DCA درمان، آن را تقریبا تمام بیماران مبتلا به زیر فانتزی یا تب تجویز می شود، حتی در صورت عدم وجود تمرکز قابل مشاهده عفونت، از آنجا که در واقع برای DCA، افزایش دمای بدن نیست معمول.
پیش بینی
مرگ و میر DCA 0.5-5٪ است، در حالی که اکثر موارد به دلیل مراقبت های پزشکی دیر و غیر متخصص است. مرگ و میر بالاترین (تا 50٪) در میان بیماران مسن تر است.
7.7.2. کما هیپروسومولار
کما هیپروسومولار(GOK) - یک عارضه حاد حاد SD-2، به دلیل کمبود آب و هیپرگلیسمی در برابر عدم وجود کمبود انسولین مطلق، همراه با مرگ و میر بالا (جدول 7.14) همراه است.
اتیولوژی
GOK، به عنوان یک قاعده، در بیماران سالخورده با SD-2 توسعه می یابد. چنین بیماران اغلب تنها هستند، بدون مراقبت، با شرایط و کنترل خودشان نادیده گرفته می شوند و مایع کافی نیستند. اغلب عفونت (سندرم پا دیابتی، پنومونی، پیلونفریت حاد)، اختلالات مغزی منجر به تخفیف می شود
گردش خون و سایر شرایط، در نتیجه بیماران به شدت در حال حرکت هستند، مواد مخدر و مایع قند را مصرف نکنید.
جدول. 7.14.Hyperosmolar Coma (GOK)
 پاتوژنز
پاتوژنز
افزایش هیپرگلیسمی و دیورزوم اسمزی، کم آبی بدن را تعیین می کند که به دلایل مشخص شده در بالا از خارج پر نشده است. نتیجه هیپرگلیسمی و کم آبی بدن، هیپروسومولاسیون پلاسما است. یک جزء جدایی ناپذیر از پاتوژنز GOK کمبود نسبی انسولین و بیش از حد هورمونهای ضد قطب است، با این وجود، که همچنان در SD-2 ادامه می یابد، ترشح باقی مانده انسولین برای سرکوب لیپولیز و کتوژنز کافی است که کتواسییدوز رخ نمی دهد.
در بعضی موارد، القاء متوسط \u200b\u200bاسیدوز ممکن است به عنوان یک نتیجه از هیپرلاکتاتاتمی در برابر پس زمینه هیپپرفوژن بافت تعیین شود. با استفاده از hyperglycemia به منظور حفظ تعادل اسمزی در مایع مغزی نخاعی، محتوای سدیم که از سلول های مغز افزایش می یابد، افزایش می یابد که پتاسیم در عوض می شود. پتانسیل ترانسفورماتور سلول های عصبی مختل شده است. روند پیشرفتی از آگاهی در ترکیب با سندرم مزمن در حال توسعه است (شکل 7.10).
همهگیرشناسی
GOK 10-30٪ از حالت های حاد هیپرگلیسمی حاد در بزرگسالان و سالمندان مبتلا به SD-2 را تشکیل می دهد. تقریبا 2/3 مورد از موارد GOK در افراد مبتلا به عدم تشخیص این دیافراگم توسعه می یابد.
تظاهرات بالینی
ویژگی های تصویر بالینی کما هیپروسومولار عبارتند از:
مجموعه ای از علائم و عوارض کم آبی بدن و هیپوفیزفوژن: تشنگی، خشکی غشاهای مخاطی، تاکی کاردی، فشار خون شریانی، تهوع، ضعف، شوک؛
تشنج های کانونی و عمومی؛
تب، تهوع و استفراغ (40-65٪ موارد)؛
از بیماری های همراه و عوارض همراه، ترومبوز رگهای عمیق اغلب یافت می شود، پنومونی، اختلالات مغزی، گاستروپارز.
تشخیصی
این بر اساس داده های تصویر بالینی، سن بیمار و سابقه SD-2، هیپرگلیسمی، در غیاب کتونوری و کتواسییدوز استوار است. علائم آزمایشگاهی معمولی GOK در جدول ارائه شده است. 7.12.
 شکل. 7 .10.
پاتوژنز کما هیپروسومولار
شکل. 7 .10.
پاتوژنز کما هیپروسومولار
تشخیص های افتراقی
سایر ایالت های حاد دیگر در بیماران مبتلا به دیابت، اغلب با آسیب شناسی همراه، منجر به عدم تمرکز ناشی از SD می شود.
رفتار
درمان و نظارت بر GOK، به استثنای برخی از ویژگی های، از آنچه که برای کما دیابتی کتوسیدیوتیک توصیف شده، متفاوت نیست (بند 7.7.1):
مقدار بیشتری از رییدر اولیه اولیه 1.5-2 لیتر در هر ساعت 1؛ 1 لیتر - برای ساعت دوم و سوم، سپس 500 میلی لیتر در ساعت محلول ایزوتونیک سدیم کلرید سدیم؛
نیاز به معرفی راه حل های حاوی پتاسیم معمولا بیشتر از کما کتواسیدوتیک است.
درمان انسولین مشابه با CC است، اما نیاز به انسولین کمتر است و سطح گلیسمی باید سریعتر از 5 mmol / l در ساعت کاهش یابد تا از توسعه ادم مغزی جلوگیری شود؛
معرفی یک راه حل هیپوتونیک (NaCl 0.45٪) بهتر است برای جلوگیری از (تنها با Hypernatremia تلفظ شده:\u003e 155 mmol / L و / یا Osmolarity کارآمد\u003e 320 MOS / L)؛
در معرفی بی کربنات هیچ نیازی نیست (فقط در بخش های احیاء تخصصی با اسیدوز با pH< 7,1).
پیش بینی
مرگ و میر در GOK بالا است و 15-60٪ است. بدترین پیش آگهی در بیماران سالمند مبتلا به آسیب شناسی شدید همراه است که اغلب علت عدم توانایی SD و توسعه GOK است.
7.7.3. هیپوگلیسمی
هیپوگلیسمی- کاهش سطح گلوکز در سرم خون (<2,2- 2,8 ммоль/л), сопровождающее клинический синдром, характеризующийся признаками активации симпатической нервной системы и/или дисфункцией центральной нервной системы. Гипогликемия как лабораторный феномен не тождественен понятию «гипогликемическая симптоматика», поскольку лабораторные данные и клиническая картина не всегда совпадают.
اتیولوژی
مصرف بیش از حد داروهای انسولین و آنالوگ های آن، و همچنین داروهای سولفونیل اوره؛
معایب غذا در زمینه شکر بدون تغییر بدون تغییر؛
پذیرش نوشیدنی های الکلی؛
اعمال فیزیکی در زمینه درمان شکر بدون تغییر و / یا بدون پذیرش کربوهیدرات اضافی؛
توسعه عوارض دیرینه SD (نوروپاتی خودمختار با گاستروپارزی، نارسایی کلیه) و تعدادی از بیماری های دیگر (شکست ناکافیان آدرنال، هیپوتیروئیدی، نارسایی کبدی، تومورهای بدخیم) با شکر بدون تغییر (ادامه پذیرش و جمع آوری TSPs در پس زمینه نارسایی کلیه، حفظ دوز انسولین سابق)؛
نقض تزریق انسولین (تزریق داخل عضلانی به جای زیر جلدی)؛
هیپوگلیسمی مصنوعی (مصرف بیش از حد داروهای قند توسط بیمار خود)؛
hyperinsulinism ارگانیک - انسولین (پاراگراف 10.3 را ببینید).
پاتوژنز
پاتوژنز هیپوگلیسمی این است که تعادل بین جریان گلوکز را به خون، دفع آن، سطح انسولین و هورمون های کنونی، نقض کند. به طور معمول، در سطح گلیسمی، در حدود 4.2-4.7 mmol / l، محصولات و انتشار انسولین از سلول های بتا سرکوب می شوند. کاهش سطح گلیسمی کمتر از 3.9 mmol / l با تحریک محصولات هورمون پیوندی (گلوکاگون، کورتیزول، هورمون رشد، آدرنالین) همراه است. علائم نوروگلیکوپنیک با کاهش سطح گلیسمی کمتر از 2.5-2.8 mmol / l توسعه می یابد. برای مصرف بیش از حد انسولینو / یا مواد مخدر سولفونیلموسیناhypoglycemia به علت اثر مستقیم هیپوگلیسیمیزه کننده یک هورمون خارجی یا اندوژن در حال توسعه است. در مورد مصرف بیش از حد توسط آماده سازی سولفونیل اوره، علائم هیپوگلیسمی ممکن است بارها پس از اتصال حمله به دلیل این واقعیت که مدت زمان دامنه داروها می تواند به روز یا بیشتر برسد، به طور مرتب تکرار می شود. TSP، که اثر تحریک کننده ای بر محصولات انسولین (متفورمین، تیاضولیدینی ها) ندارد، خود هیپوگلیسمی خود را نمی توان به تنهایی ایجاد کرد، اما زمانی که سولفونیل اوره یا انسولین را اضافه می کنند، دریافت دومی در همان دوز ممکن است باعث ایجاد هیپوگلیسمی ناشی از آن شود جمع آوری اثر شاهزاده از درمان ترکیبی (جدول 7.15).
جدول. 7.15.هیپوگلیسمی
 پایان جدول 7.15
پایان جدول 7.15
 هنگام دریافت الکلاین اتفاق می افتد سرکوب گلوکگنز در کبد، که مهمترین عامل هیپوگلیسمی است. تمرین فیزیکیاز استفاده از گلوکز وابسته به انسولین محافظت کنید، به این دلیل که علل هیپوگلیسمی در زمینه درمان بدون شکر بدون شکر و / یا در صورت عدم پذیرش اضافی کربوهیدرات وجود دارد.
هنگام دریافت الکلاین اتفاق می افتد سرکوب گلوکگنز در کبد، که مهمترین عامل هیپوگلیسمی است. تمرین فیزیکیاز استفاده از گلوکز وابسته به انسولین محافظت کنید، به این دلیل که علل هیپوگلیسمی در زمینه درمان بدون شکر بدون شکر و / یا در صورت عدم پذیرش اضافی کربوهیدرات وجود دارد.
همهگیرشناسی
نور، به سرعت هیپوگلیسمی حباب در بیماران مبتلا به SD-1، دریافت درمان انسولین شدید، می تواند چندین بار در هفته و نسبتا بی ضرر توسعه یابد. در هر بیمار که در درمان انسولین شدید است، در هر سال به مدت 1 مورد هیپوگلیسمی شدید است. در اغلب موارد، هیپوگلیسمی در شب در حال توسعه است. با SD-2 در 20٪ از بیماران دریافت کننده انسولین، و در 6٪ دریافت آماده سازی سولفونیلوروری، برای حداقل یک قسمت از هیپوگلیسمی شدید برای حداقل یک قسمت توسعه می یابد.
تظاهرات بالینی
دو گروه اصلی علائم متمایز هستند: آدرنرژیک، همراه با فعال شدن سیستم عصبی سمپاتیک و انتشار آدرنالین آدرنالین و نوروگلیکوپنیک، همراه با عملکرد اختلالات سیستم عصبی مرکزی در برابر پس زمینه کمبود زیرمجموعه انرژی اصلی آن است. به آدرنرژیکعلائم عبارتند از: تاکیکاردی، MyDriasis؛ اضطراب، پرخاشگری؛ لرزش، عرق سرد، پارستزی؛ تهوع، گرسنگی قوی، perspersalization؛ اسهال، ادرار فراوان. به نوروگلیکوپنیعلائم عبارتند از: آستنیا،
کاهش غلظت توجه، سردرد، احساس ترس، سردرگمی، بی نظمی، توهم؛ سخنرانی، اختلالات بصری، اختلالات رفتاری، آمنیازیا، نقض آگاهی، تشنج، فلج گذرا، به آنها. وابستگی واضح به شدت و دنباله ای از توسعه علائم به عنوان هیپوگلیسمی ممکن نیست. تنها علائم نروژلیکوپنی تنها ممکن است رخ دهد. در بعضی موارد، علیرغم بازسازی نوروگلیسمی و درمان مداوم، بیماران می توانند در یک حالت کشویی یا حتی کمیته بیش از چند ساعت و حتی روز باشند. هیپوگلیسمی بلند مدت یا قسمت های مکرر آن می تواند منجر به تغییرات غیر قابل برگشت در سیستم عصبی مرکزی (در درجه اول در قشر نیمکره های بزرگ)، تظاهرات آن به طور قابل توجهی از قسمت های خوشمزه و توهم پارانوئید به تشنج های صرع معمولی متفاوت است، که این است نتیجه اجتناب ناپذیر که از زوال عقل مقاوم هستند.
هیپرگلیسمی به لحاظ ذهنی به بیماران سبک تر از اپیزود ها حتی هیپوگلیسمی نور منتقل می شود. بنابراین، بسیاری از بیماران به علت ترس از هیپوگلیسمی، لازم است که گلیسمی را در سطح نسبتا بالا حفظ کنند، که در واقع مربوط به عدم توانایی بیماری است. غلبه بر این کلیشه نیازمند تلاش های قابل توجهی از پزشکان و پرسنل آموزشی است.
تشخیصی
تصویر بالینی هیپوگلیسمی در یک بیمار مبتلا به SD در ترکیب با آزمایشگاه (به عنوان یک قاعده، با گلوکز متر) تشخیص سطح قند خون کم.
تشخیص های افتراقی
دلایل دیگر منجر به از دست دادن آگاهی می شود. اگر علت از دست دادن آگاهی از بیمار SD ناشناخته باشد، غیر ممکن است تجزیه و تحلیل بیان سطح گلیسمی، آن را نشان می دهد معرفی گلوکز. اغلب نیاز به پیدا کردن علل توسعه هیپوگلیسمی مکرر در بیماران مبتلا به SD وجود دارد. اغلب آنها نتیجه درمان ناکافی شکر و دانش بیمار پایین تر از بیماری آنها هستند. لازم به ذکر است که برای کاهش نیاز به درمان قند تا زمانی که لغو کامل آن ("SD ناپدید شده) می تواند تعدادی از بیماری ها (نارسایی آدرنال، کم کاری تیروئید، نارسایی کلیوی و کبد) را شامل شود، از جمله تومورهای بدخیم.
رفتار
برای درمان هیپوگلیسمی نور، که در آن بیمار آگاه است و ممکن است به خود کمک کند، معمولا به اندازه کافی برای مصرف غذا یا مایع حاوی کربوهیدرات ها در مقدار 1-2 واحد نان (10 تا 20 گرم گلوکز) کافی است. چنین تعداد شامل، به عنوان مثال، در 200 میلی لیتر آب میوه شیرین است. نوشیدنی ها به طور موثر از هیپوگلیسمی جلوگیری می کنند، زیرا در شکل مایع گلوکز به طور قابل توجهی جذب می شود. اگر علائم همچنان رشد کند، به رغم ادامه پذیرش کربوهیدرات ها، لازم است تزریق داخل وریدی گلوکون یا گلوکاگون عضلانی باشد. به همان شیوه، هیپوگلیسمی شدید با از دست دادن آگاهی درمان می شود. در این مورد، بیمار حدود 50 میلی لیتر معرفی شده است 40٪ محلول گلوکز داخل وریدی.معرفی گلوکز باید تا زمانی که اتصال حمله و عادی سازی گلیسمی ادامه یابد، همچنان ادامه دارد، هرچند دوز بیشتر تا 100 میلی لیتر و بیشتر به عنوان یک قاعده، مورد نیاز نیست. گلوکگوناین معرفی شده است (به عنوان یک قاعده، تهیه شده در شرایط کارخانه پر از یک سرنگ) به صورت عضلانی یا زیر جلدی. پس از چند دقیقه، سطح گلیقیم به دلیل القاء گلوکولولیز گلوکاگون، نرمال شده است. با این حال، این همیشه اتفاق نمی افتد: با سطح بالایی از انسولین در گلوکاگون خون بی اثر است. نیمه عمر گلوکاگون کوتاهتر از انسولین است. با الکل و بیماری های کبدی، سنتز گلیکوژن مختل می شود و تزریق گلوکاگون می تواند بی اثر باشد. یک اثر جانبی از تجویز گلوکاگون ممکن است استفراغ باشد که خطر آسپیراسیون را ایجاد می کند. بیمار نزدیک بیمار ترجیحا تکنیک تزریق گلوکاگون را ترجیح می دهد.
پیش بینی
هیپوگلیسمی نور در بیماران آموزش دیده در برابر پس زمینه جبران خسارت بیماری خوب، ایمن است. هیپوگلیسمی مکرر نشانه ای از جبران خسارت SD ضعیف است؛ در اغلب موارد، چنین بیماران در طول روز بقیه روز روز با هیپرگلیسمی بیشتر یا کمتر مشخص شده و سطح بالایی از هموگلوبین گلیکوز شده تعیین می شود. بیماران سالمند مبتلا به عوارض دیررس هیپوگلیسمی SD می توانند چنین عوارض عروقی را به عنوان انفارکتوس میوکارد، سکته مغزی، خونریزی شبکیه ای تحریک کنند. کما هیپوگلیسمیک به مدت 30 دقیقه با درمان مناسب و بازگشت سریع آگاهی، به عنوان یک قاعده، عوارض و عواقب آن را ندارد.
7.8. عوارض دیررس دیابت
عوارض دیررس در حال توسعه با هر دو نوع SD هستند. به صورت بالینی پنج عوارض اصلی اولیه SD را تخصیص می دهد: ماکرونگیاپاتی، نفروپاتی، رتینوپاتی، نوروپاتی و سندرم پا دیابتی. غنی از عوارض دیررس برای انواع مختلف SD به وسیله این واقعیت تعیین می شود که پیوند پاتوژنیک اصلی آنها هیپرگلیسمی مزمن است. در این راستا، در زمان تظاهرات SD-1، عوارض دیررس در بیماران تقریبا هرگز ملاقات، توسعه از طریق سالها و دهه ها، بسته به اثربخشی درمان. بزرگترین ارزش بالینی در SD-1، به عنوان یک قاعده، به دست آوردن میکروآنژیوپاتی دیابتی(نوروپاتی، رتینوپاتی) و نوروپاتی (سندرم پا دیابتی). با SD-2، برعکس، عوارض دیررس اغلب در زمان تشخیص تشخیص داده می شود. اول، این به دلیل این واقعیت است که SD-2 طولانی قبل از تشخیص ایجاد می شود. در مرحله دوم، آترواسکلروز، ماکرونگتیوپاتی بالینی ظاهر می شود، دارای پاتوژنز زیادی با SD است. با SD-2، بزرگترین اهمیت بالینی، به عنوان یک قاعده، دیابتی را به دست می آورد macroangiopathy،که در زمان تشخیص از اکثریت قریب به اتفاق بیماران تشخیص داده می شود. در هر مورد خاص، مجموعه و شدت عوارض دیرین فرد از عدم وجود پارادوکسیک کامل آنها متفاوت است، به رغم مدت زمان قابل توجهی از بیماری تا ترکیبی از تمام گزینه های ممکن در فرم سنگین.
عوارض دیررس هستند علت اصلی مرگبیماران مبتلا به SD و با توجه به شیوع آن - مهمترین مسئله پزشکی و اجتماعی مراقبت های بهداشتی اکثر کشورها. مربوط به هدف اصلی درمانو مشاهدات بیماران مبتلا به CD پیشگیری (اولیه، ثانویه، ثانویه، ثانویه) عوارض دیررس آن است.
7.8.1 Macroangiopathy دیابتی
Macroangiopathy دیابتی- یک مفهوم جمعی که ضایعه آترواسکلروز از شریان های بزرگ را در SD ترکیب می کند،
به طور بالقوه از طریق بیماری های قلبی ایسکمیک (IBS)، آترواسکلروز مورب، عروق مغزی، اندام های پایین تر، اندام های داخلی و فشار خون شریانی (جدول 7.16) ظاهر می شود.
جدول. 7.16.Macroangiopathy دیابتی
 علت و پاتوژنز
علت و پاتوژنز
احتمالا شبیه به علت و پاتوژنز آترواسکلروز در افراد بدون SD است. پلاک های آترواسکلروز در ساختار میکروسکوپی در افراد SD و بدون آن متفاوت نیست. با این وجود، با SD به پیش زمینه، عوامل خطر اضافی می تواند انجام شود، یا SD عوامل شناخته شده غیر اختصاصی را تشدید می کند. به همین ترتیب، SD باید شامل موارد زیر باشد:
1. هیپرگلیسمی.این یک عامل خطر برای آترواسکلروز است. افزایش سطح HbA1c با 1٪ در بیماران مبتلا به SD-2 افزایش می یابد
خطر ابتلا به انفارکتوس میوکارد 15٪. مکانیسم عمل آتروژنیک هیپرگلیسمی کاملا روشن نیست، ممکن است آن را با گلیکوزینگ محصولات نهایی متابولیسم LDL و کلاژن دیواره عروقی همراه باشد.
2. فشار خون شریانی(AG). در پاتوژنز، اهمیت زیادی به مولفه کلیه متصل می شود (نفروپاتی دیابتی).AG با SD-2 - هیچ گونه خطر ابتلا به انفارکتوس و سکته مغزی کمتر از هیپرگلیسمی نیست.
3. دیسلیپیدمیHyperinsulamia، که یک جزء غیر قابل انعطاف از مقاومت به انسولین در SD-2 است، باعث کاهش سطح HDL می شود، افزایش سطح تری گلیسیرید و کاهش تراکم، I.E. تقویت LDL آتروژنیک.
4. چاقیکه اکثر بیماران مبتلا به SD-2 رنج می برند، یک عامل مستقل خطر ابتلا به آترواسکلروز، انفارکتوس میوکارد و سکته مغزی است (به بند 11.2 مراجعه کنید).
5. مقاومت به انسولین.Hyperinsulamia و سطوح بالای مولکول های انسولین مانند پروانهلین، خطر ابتلا به آترواسکلروز را افزایش می دهد که ممکن است با اختلال عملکرد اندوتلیال همراه باشد.
6. اختلال انعقاد خون.در SD، افزایش سطح فیبرینوژن، فعال کننده مهار کننده پلاکت و عامل Willebrand، به عنوان یک نتیجه از آن حالت پروترومبوتیک از سیستم خون انعطاف پذیر تشکیل شده است.
7. اختلال عملکرد اندوتلیالبا افزایش بیان فعال کننده مهار کننده پلاسمینوژن و مولکول های چسبندگی سلولی مشخص می شود.
8. استرس اکسیداتیومنجر به افزایش غلظت LDL اکسید شده و F2-ISO-Perts می شود.
9. التهاب سیستمیککه در آن بیان پروتئین فیبرینوژن و C-واکسیناسیون C افزایش می یابد.
مهمترین عوامل خطر ابتلا به IWC در SD-2 سطح بالایی از LDL، کاهش HDL، فشار خون شریانی، هیپرگلیسمی و سیگار کشیدن است. یکی از تفاوت های فرایند آترواسکلروز در SD شایع تر است ماهیت ديستال ضایعات اکلوزال،کسانی که. این فرآیند اغلب در شریان های نسبتا کوچکتر دخالت می کند، که باعث درمان جراحی و پیش بینی می شود.
همهگیرشناسی
خطر ابتلا به IBS در افراد مبتلا به SD-2 6 برابر بیشتر از افراد بدون دیابت، در حالی که برای مردان و زنان یکسان است. پرفشاری خون شریانی در 20٪ بیماران مبتلا به SD-1 و 75٪ از SD-2 تشخیص داده می شود. به طور کلی، در بیماران مبتلا به SD، 2 برابر بیشتر از آنهایی که بدون او هستند، ملاقات می کنند. آترواسکلروز Obricolary از عروق محیطی در 10٪ بیماران مبتلا به SD در حال توسعه است. ترومبوآمبولی عروق مغزی در 8٪ بیماران مبتلا به SD (2-4 برابر بیشتر از افراد بدون SD) رشد می کند.
تظاهرات بالینی
اغلب از کسانی که بدون SD نیستند متفاوت نیستند. در تصویر بالینی عوارض Macrovascular SD-2 (انفارکتوس میوکارد، سکته مغزی، آسیب انسداد به عروق پا) اغلب پیروز می شود و دقیقا برای اولین بار یک هیپرگلیسمی در توسعه آنها در یک بیمار است. شاید به علت نوروپاتی مستقل خودمختار تا 30 درصد انفارکتوس میوکارد در افراد مبتلا به جریان سی دی بدون حمله آنژیوسی معمولی (انفارکتوس اداری).
تشخیصی
اصول تشخیص عوارض آترواسکلروز (IBS، اختلال مغزی مغزی، آسیب انسداد به شریان های پاها) از افراد بدون SD متفاوت نیست. اندازه گرفتن فشار شریانی(AD) باید در هر بازدید از بیمار با SD به دکتر، و تعریف شاخص ها انجام شود طیف لیپیدخون (کلسترول تام، تری گلیسیرید، LDL، HDL) در SD باید حداقل یک بار در سال انجام شود.
تشخیص های افتراقی
سایر بیماری های قلبی عروقی، پرفشاری خون شریانی علائم، دیمپیدمی ثانویه.
رفتار
♦ کنترل فشار خون.سطح مناسب فشار خون سیستولیک در SD کمتر از 130 میلی متر جیوه و دیاستولیک 80 mmng (جدول 7.3) است. اکثر بیماران برای رسیدن به این هدف نیاز به داروهای هیپوتوقی دارند. داروهایی برای انتخاب درمان هیپوتیزم در Cd، مهار کننده های ACE و مسدود کننده های گیرنده آنژیوتانسین هستند که در صورت لزوم، دیورتیک های تیازید تکمیل می شوند. آماده سازی انتخاب برای بیماران مبتلا به SDS که تحت انفارکتوس میوکارد قرار گرفته اند، β-adrenoblays هستند.
♦ اصلاح ناسازگاری.سطوح هدف از شاخص های طیف لیپید در جدول ارائه شده است. 7.3. آماده سازی انتخاب درمان هیپولیپیدمی مهارکننده های 3-هیدروکسی -3-متیل گلوله-co-reductase (استاتین ها) است.
♦ درمان ضد عفونی کننده.درمان آسپرین (75-100 میلی گرم در روز) به بیماران مبتلا به SD مبتلا به SD بیش از 40 سال با افزایش خطر ابتلا به پاتولوژی قلبی عروقی (سابقه خانوادگی، فشار خون بالا، سیگار کشیدن، دیس لیپیدمی، میکروآلبومینوری) و همچنین همه بیماران نشان داده شده است با تظاهرات بالینی آترواسکلروز به عنوان پیشگیری ثانویه.
♦ غربالگری و درمان IBS.آزمایشات بار برای محرومیت از IBS به بیماران مبتلا به علائم بیماری های قلبی عروقی، و همچنین شناسایی آسیب شناسی در ECG نشان داده شده است.
پیش بینی
75٪ بیماران مبتلا به SD-2 و 35٪ بیماران مبتلا به SD-1 از بیماری های قلبی عروقی می میرند. تقریبا 50٪ از بیماران مبتلا به SD-2 از عوارض CHD میمیرند، 15٪ از ترومبوآمبولیک عروق مغزی. مرگ و میر از انفارکتوس میوکارد در افراد مبتلا به SD بیش از 50٪ است.
7.8.2. رتینوپاتی دیابتی
رتینوپاتی دیابتی(DR) - میکروآنژیوپاتی از عروق شبکیه چشم، مشخص شده توسط توسعه میکرو محیط زیست، خونریزی، تغییرات اگزودیتی و تکثیر عروق تازه تشکیل شده، منجر به از دست دادن جزئی یا کامل از بین رفتن دید (جدول 7.17).
اتیولوژی
عامل اصلی علمی در توسعه DR هیپرگلیسمی مزمن است. عوامل دیگر (پرفشاری خون شریانی، دیس لیپیدمی، سیگار کشیدن، بارداری، و غیره) اهمیت کمتری دارند.
پاتوژنز
پیوندهای اصلی پاتوژنز DR عبارتند از:
Microangiopathy رگ های شبکه، منجر به محدود شدن نظارت بر عروق با توسعه هیپپرفوژن؛
دژنراسیون عروق با تشکیل میکروسکوپ؛
هیپوکسی پیشرفته، تحریک تکثیر عروق و منجر به دیستروفی چربی و رسوب نمک های کلسیم در شبکیه؛
جدول. 7.17.رتینوپاتی دیابتی
 MicroIndarcts با Exudation، منجر به تشکیل نرم "نقاط پنبه"؛
MicroIndarcts با Exudation، منجر به تشکیل نرم "نقاط پنبه"؛
رسوب لیپید ها با تشکیل اگزودای های متراکم؛
رشد شبکیه از عروق های تکثیر کننده برای ایجاد شانزدها و آنوریسم، منجر به انقباض رگ ها و تشدید هیپپرفوژن شبکیه می شود؛
پدیده اعتماد به پیشرفت بیشتر ایسکمیک، که علت تشکیل نفوذ و زخم ها است؛
جداسازی شبکیه به عنوان یک نتیجه از تجزیه ایسکمیک آن و شکل گیری intreoretinal intreoretinal؛
خونریزی در بدن شیشه ای به عنوان یک نتیجه از حملات قلب هموراژیک، تهاجم عروقی عظیم و آنوریسم رگ؛
تکثیر عروق عنبیه (روبل دیابتی)، منجر به توسعه گلوکوم ثانویه می شود؛
ماکولوپاتی با تورم تورم.
همهگیرشناسی
دیگر شایع ترین علت نابینایی در میان جمعیت کار کشورهای توسعه یافته و خطر توسعه نابینایی در بیماران مبتلا به دیابت 10-20 برابر بیشتر از جمعیت کلی است. در زمان تشخیص SD-1، DR توسط تقریبا هر یک از بیماران تشخیص داده نمی شود، پس از 5 سال، بیماری در 8٪ از بیماران شناسایی شده و با یک دوره سی سال دیابت - در 98٪ از بیماران. در زمان تشخیص، SD-2 DR در 20-40٪ بیماران و در میان بیماران مبتلا به پانزده سال تجربه، SD-2 - در 85٪ تشخیص داده می شود. در SD-1، رتینوپاتی پرولیفراتیو نسبتا بیشتر بیشتر و در SD-2 - ماکولوپاتی (75٪ موارد ماکولوپاتی) مربوط می شود.
تظاهرات بالینی
با توجه به طبقه بندی به طور کلی پذیرفته شده، 3 مرحله از DR ها متمایز هستند
(جدول 7.18).
تشخیصی
معاینه چشم پزشکی کامل، که شامل Ophthalmoscopy مستقیم با عکاسی از شبکیه، به بیماران مبتلا به SD-1 پس از 3-5 سال پس از تظاهرات بیماری نشان داده شده است و بیماران مبتلا به SD-2 بلافاصله پس از آن تشخیص داده می شود. در آینده، چنین تحقیقاتی باید سالانه تکرار شود.
جدول. 7.18.طبقه بندی رتینوپاتی دیابتی
 تشخیص های افتراقی
تشخیص های افتراقی
سایر بیماری های چشم در بیماران مبتلا به دیابت.
رفتار
اصل اساسی درمان رتینوپاتی دیابتی، و همچنین سایر عوارض دیررس، جبران مطلوب SD است. موثرترین روش درمان رتینوپاتی دیابتی و جلوگیری از نابینایی است فوتوکوئوگولاسیون لیزر.هدف
 شکل. 7.11.رتینوپاتی دیابتی:
شکل. 7.11.رتینوپاتی دیابتی:
الف) غیر پرولیفراتیو؛ ب) prepolytematic؛ ج) proliferative
فوتوکوئوگولاسیون لیزر، پایان دادن به عملکرد عروق تازه تشکیل شده است که نشان دهنده تهدید اصلی برای توسعه چنین عوارض سنگین مانند هموفتلم، جداسازی شبکیه کششی، ریزه شبکیه، ریزش عنبیه و گلوکوم ثانویه است.
پیش بینی
نابینایی در 2٪ بیماران مبتلا به CD (3-4٪ بیماران مبتلا به SD-1 و 1.5-2٪ بیماران مبتلا به SD-2) ثبت می شود. فراوانی تقریبی موارد جدید نابینایی مرتبط با DR 3.3 مورد در هر 100،000 جمعیت در هر سال است. در طی SD-1، کاهش HbA1C به 7.0٪ منجر به کاهش خطر ابتلا به DR توسعه 75٪ و کاهش خطر ابتلا به 60٪ کاهش می یابد. در SD-2، کاهش HbA1c به میزان 1٪ منجر به کاهش خطر ابتلا به DR توسعه 20٪ می شود.
7.8.3. نفروپاتی دیابتی
نفروپاتی دیابتی(DNF) به عنوان آلبومینوری (بیش از 300 میلی گرم آلبومین در روز یا پروتئینوریوم بیش از 0.5 گرم پروتئین در روز) تعریف می شود و / یا کاهش عملکرد فیلتر کلیه ها در افراد مبتلا به CD در غیاب عفونت های ادراری نارسایی قلبی یا سایر بیماری های کلیوی. میکروآلبومینوریا به عنوان دفع آلبومین 30-300 میلی گرم در روز یا 20-200 میکروگرم در دقیقه تعریف می شود.
علت و پاتوژنز
عوامل خطر اصلی DNF طول مدت SD، هیپرگلیسمی مزمن، پرفشاری خون شریانی، دیس لیپیدمی، بیماری کلیوی از والدین است. هنگامی که DNF عمدتا تحت تاثیر قرار می گیرد دستگاه خوشه ایکلیه
1. یکی از مکانیسم های ممکن برای آن هیپرگلیسمیترویج توسعه ضایعه گلومرها، انباشت سوربیتول به علت فعال شدن مسیر پلی اتیل متابولیسم گلوکز، و همچنین تعدادی از محصولات Gyricing محدود است.
2. نقض های همودینامیک، یعنی پرفشاری خون شریانی introlubok(افزایش فشار خون در داخل گلوی کلیه) مهمترین مولفه پاتوژنز است
علت فشار خون داخل مغزی نقض تن از آرتریول است: گسترش نتیجه و محدود شدن پایان.
جدول. 7.19.نفروپاتی دیابتی
 این، به نوبه خود، تحت تاثیر تعدادی از عوامل هومورال، مانند آنژیوتانسین -2 و اندوتلین، و همچنین به دلیل نقض خواص الکترولیت غشای پایه گلومرول رخ می دهد. علاوه بر این، فشار خون داخل مغزی به فشار خون سیستمیک کمک می کند، که توسط اکثر بیماران مبتلا به DNF تعیین می شود. با توجه به فشار خون داخل مغزی، آسیب به غشاهای بازال و منافذ فیلتراسیون وجود دارد
این، به نوبه خود، تحت تاثیر تعدادی از عوامل هومورال، مانند آنژیوتانسین -2 و اندوتلین، و همچنین به دلیل نقض خواص الکترولیت غشای پایه گلومرول رخ می دهد. علاوه بر این، فشار خون داخل مغزی به فشار خون سیستمیک کمک می کند، که توسط اکثر بیماران مبتلا به DNF تعیین می شود. با توجه به فشار خون داخل مغزی، آسیب به غشاهای بازال و منافذ فیلتراسیون وجود دارد
از طریق آن آهنگ ها شروع به نفوذ می کنند (میکروآلبومینوری)،و سپس مقدار قابل توجهی از آلبومین (پروتئینوری).ضخیم شدن غشاهای بازال باعث تغییر در خواص الکترولیت آنها می شود که به خودی خود منجر به مقدار بیشتری از آلبومین در اولترافیلترات حتی در صورت عدم تغییر اندازه منافذ فیلتراسیون می شود.
3. استعداد ژنتیکی.بستگان بیماران مبتلا به DNF با افزایش فرکانس افزایش فشار خون شریانی. داده های مربوط به اتصال DNF با پلی مورفیسم ژن ACE وجود دارد. میکروسکوپیک، در طول DNF، ضخیم شدن غشای پایه گلومرها، گسترش mezanygia، و همچنین تغییرات فیبری که باعث ایجاد آرتريول ها می شود، نشان می دهد. در مرحله نهایی، که به صورت بالینی به نارسایی مزمن کلیوی (CPN) مربوط می شود، کانونی (Kimmelistil-Wilson) تعیین می شود و سپس گلومرولروز را منتشر می کند.
همهگیرشناسی
میکروآلبومینوریا در 6-60٪ از بیماران مبتلا به SD-1 5-15 سال پس از تظاهرات آن تعیین می شود. DNF 35٪ از SD-1 تعیین می شود، اغلب در مردان و افرادی که در سن 15 سالگی توسعه یافته اند. در SD-2، DNF در 25 درصد از نمایندگان نژاد اروپایی و 50 درصد از نژاد آسیایی توسعه می یابد. شیوع کلی DNF در SD-2 4-30٪ است.
تظاهرات بالینی
یک تظاهرات بالینی نسبتا اولیه، که به طور غیر مستقیم با DNF متصل است، فشار خون شریانی است. سایر تظاهرات صریح بالینی متعلق به اواخر است. این شامل تظاهرات سندرم نفروتیک و نارسایی مزمن کلیوی است.
تشخیصی
غربالگری به DNF در افراد مبتلا به CD نشان می دهد که آزمایش سالانه microalbuminuriaبا SD-1 5 سال پس از تظاهرات بیماری و در SD-2 بلافاصله پس از آن شناسایی شد. علاوه بر این، حداقل یک تعریف سالانه از سطوح کراتینین برای محاسبه ضروری است سرعت فیلتراسیون گلومرولی (SCF).به عنوان مثال، SCF را می توان با استفاده از فرمول های مختلف محاسبه کرد، به عنوان مثال، توسط فرمول Courft-Golta:
برای مردان: A \u003d 1،23 (NOP از SCF 100 - 150 میلی لیتر در دقیقه) برای زنان: A \u003d 1.05 (هنجار SCF 85 - 130 میلی لیتر در دقیقه)
در مراحل اولیه DNF، افزایش SCF می تواند نشان داده شود که به تدریج به عنوان CPN توسعه می یابد. میکروآلبومینوری آغاز می شود 5-15 سال پس از تظاهرات SD-1 تعیین می شود. با SD-2 در 8-10٪ موارد، بلافاصله پس از آن تشخیص داده می شود، احتمالا به دلیل یک دوره طولانی بدون علامت از بیماری تا تشخیص. اوج توسعه پروتئینوری صریح یا آلبومینوری در SD-1 بین 15 تا 20 بعد از شروع آن کاهش می یابد. پروتئینوری می گوید: برگشت ناپذیرDNF، که دیر یا زود به CPN منجر می شود. اورمیا به طور متوسط \u200b\u200b7-10 سال پس از ظهور پروتئینوری صریح است. لازم به ذکر است که SCF با پروتئینوری ارتباط ندارد.
تشخیص های افتراقی
سایر علل پروتئینوری و نارسایی کلیوی در افراد SD. در اغلب موارد، DNF با فشار خون بالا، رتینوپاتی دیابتی یا نوروپاتی ترکیب شده است، در صورت عدم وجود تشخیص افتراقی باید کاملا کامل باشد. در 10٪ موارد، با SD-1 و در 30٪ موارد، در طی SD-2، پروتئینوری مربوط به DNF نیست.
رفتار
♦ شرایط اصلی اولیه و ثانویه جلوگیری
dnfجبران SD و حفظ فشار خون طبیعی سیستمیک هستند. علاوه بر این، پیشگیری اولیه از DNF، کاهش مصرف مواد غذایی پروتئین را کاهش می دهد - کمتر از 35٪ کالراژ روزانه.
♦ در مراحل microalbuminuriaو پروتئینوریبیماران نشان می دهد انتصاب مهارکننده های ACE یا مسدود کننده های گیرنده آنژیوتانسین. با فشار خون بالا، آنها در دوزهای هیپوتیزم تجویز می شوند، در صورت لزوم در ترکیب با سایر داروهای هیپنواخت. در فشار شریانی طبیعی، این داروها در دوزهای تجویز تجویز می شوند که منجر به توسعه هیپوتانسیون نمی شود. هر دو مهار کننده های ACE (در SD-1 و SD-2) و مسدود کننده های گیرنده آنژیوتانسین (در SD-2) به جلوگیری از انتقال میکروآلبومینوری به پروتئینوری کمک می کنند. در بعضی موارد، در برابر پس زمینه این درمان، در ترکیب با جبران دیابت برای پارامترهای دیگر، میکروآلبومینوری حذف می شود. علاوه بر این، شروع از مرحله میکروآلبومینوری ضروری است
کاهش مصرف پروتئین ها کمتر از 10٪ کالری روزانه (یا کمتر از 0.8 گرم در کیلوگرم وزن) و نمک کمتر از 3 گرم در روز است.
♦ در مرحله cpnبه عنوان یک قانون، تصحیح درمان شکر مورد نیاز است. اکثر بیماران مبتلا به SD-2 باید به درمان انسولین ترجمه شوند، زیرا Cumulation TSP خطر ابتلا به هیپوگلیسمی شدید را حمل می کند. اکثر بیماران مبتلا به SD-1 کاهش نیاز به انسولین را کاهش می دهند، زیرا کلیه یکی از مهمترین مکان های متابولیسم آن است. با افزایش سطح سرمی کراتینین به 500 میکرومول بر لیتر و نیاز بیشتری به مطرح شدن سوال از آماده سازی بیمار به روش تجمع خارجی (همودیالیز، دیالیز دیالیز) یا جراحی (پیوند کلیه). پیوند کلیه در سطوح کراتینین تا 600-700 μmol / L نشان داده شده و سرعت فیلتراسیون گلومرولی کمتر از 25 میلی لیتر در دقیقه، همودیالیز - 1000-1200 μmol / L و کمتر از 10 میلی لیتر در دقیقه کاهش می یابد.
پیش بینی
در 50٪ از بیماران مبتلا به SD-1 و 10٪ با SD-2، که پروتئینوری را تشخیص می دهند، CPN در طی 10 سال آینده توسعه می یابد. 15٪ از تمام مرگ و میر بیمار مبتلا به SD-1 در سن 50 سالگی با CPN به علت DNF همراه است.
7.8.4 نوروپاتی دیابتی
نوروپاتی دیابتی(DN) ترکیبی از سندرم های ضایعات سیستم عصبی است که می تواند بسته به دخالت شایع در فرآیند ادارات مختلف آن (Sensorny، خودمختار)، و همچنین شیوع و شدت ضایعه (جدول 7.20) طبقه بندی شود.
من. نوروپاتی Sensomotor:
متقارن؛
کانونی (mononeropathy) یا پلیفیک (جمجمه، موتور پروگزیمال، اندام های مونونروئید و لگن).
دوم نوروپاتی خودمختار (رویشی):
قلب و عروق (هیپوتانسیون ارتوپداتیک، سندرم Denervation قلب)؛
دستگاه گوارش (معده Atony، Dyskinesia از دستگاه صفراوی، انتروپاتی دیابتی)؛
Urogenital (با اختلال عملکرد مثانه و عملکرد جنسی)؛
نقض در توانایی بیمار برای تشخیص هیپوگلیسمی؛
نقض عملکرد دانش آموز؛
نقض عملکرد غدد عرق (آنیدروز دفاعی، هیپیدروز در هنگام خوردن).
جدول. 7.20.نوروپاتی دیابتی
 علت و پاتوژنز
علت و پاتوژنز
علت اصلی روز هیپرگلیسمی است. مکانیسم های مختلف پاتوژنز آن فرض می شود:
فعال سازی مسیر پلیول از متابولیسم گلوکز، به عنوان یک نتیجه از آن سوربیتول، فروکتوز و کاهش mioindose و گلوتاتیون در سلول های عصبی رخ می دهد. این، به نوبه خود منجر به فعال شدن فرآیندهای رادیکال آزاد و کاهش سطح اکسید نیتروژن می شود؛
گلیکوزیل شدن غیر آنزیمی پروتئین های غشایی و سیتوپلاسمی سلول های عصبی؛
ميکرونگيوپاتی vasa nervorum،که منجر به کاهش سرعت جریان خون مویرگی و هیپوکسی عصبی می شود.
همهگیرشناسی
شیوع پایین با هر دو نوع SD حدود 30٪ است. با SD-1 5 سال بعد، شروع به تشخیص 10٪ از بیماران می شود. فراوانی موارد جدید پایین در SD-2 حدود 6٪ از بیماران در سال است. بیشترین گزینه، روز حساسیت متقارن دیستال است.
تظاهرات بالینی
Sensomotor DN.توسط یک مجموعه ای از اختلالات حرکتی و حساس ظاهر می شود. علائم مکرر فرم ديستال پارستزیکه خود را با احساس "خزنده گوزن ها"، بی حسی، آشکار می کند. بیماران اغلب در مورد سر پا شکایت می کنند، هرچند که آنها به لمس گرم می شوند، که نشانه ای است که اجازه می دهد تا پلینیوپاتی را از تغییرات ایسکمیک تشخیص دهد، زمانی که پا روی لمس سرد است. تظاهرات اولیه نوروپاتی حسی، نقض حساسیت ارتعاش است. مشخصه سندرم "پا بی سر و صدا" است که ترکیبی از پارستزی شبانه و حساسیت بالا است. درد در پاهااغلب در شب نگران هستند، در حالی که گاهی اوقات بیمار می تواند لمس پتو را از بین ببرد. در یک مورد معمولی درد، به عنوان مخالف با چنین بیماری های نابالغ، شریان ها می توانند هنگام راه رفتن کاهش یابد. پس از سالها، درد می تواند به صورت خود به خودی به علت مرگ فیبرهای عصبی کوچک مسئول حساسیت درد، خاتمه یابد. وابسته به هیپوستشیاین توسط از دست دادن حساسیت توسط نوع "جوراب" و "دستکش" ظاهر می شود. نقض حساسیت عمیق و پروپریپسی، منجر به نقض هماهنگی و مشکل حرکت (Ataxia حسی) می شود. بیمار در مورد "پاهای دیگر" شکایت می کند، احساس "ایستادن بر روی پنبه". نقض انسداد تروفیک منجر به تغییرات دژنراتیو در پوست، استخوان ها و تاندون ها می شود. نقض حساسیت درد منجر به مکرر، نه یک بیمار قابل توجه با توقف microtraummamm که به راحتی آلوده است. نقض هماهنگی و راه رفتن منجر به توزیع مجدد فیزیولوژیکی بار در مفاصل پا می شود. در نتیجه، یک رابطه تشریحی در دستگاه واژینال پاها مختل می شود.
قوس پای تغییر شکل، تورم، شکستگی ها، فرآیندهای مزمن فسخ است (به بند 7.8.5 مراجعه کنید).
چندین فرم از روز خودمختار. علت فرم قلب و عروق- اختلال ناشی از انسداد قلب و عروق و عروق بزرگ. عصب سرگردان طولانی ترین عصب است، در ارتباط با آن قبل از دیگران شگفت زده شده است. به عنوان یک نتیجه از غلبه بر تأثیرات سمپاتیک توسعه می یابد استراحت Tahcardiaپاسخ ناکافی به Orthostasis خود را نشان می دهد هیپوتانون Ortostaticو حالت های Syncopal. انزوای گیاهی از مجموعه پیچیده ریه منجر به عدم وجود تغییرات ریتم قلب می شود. با نوروپاتی خودمختار، افزایش شیوع در بیماران مبتلا به Neckcades Miocardial SD مرتبط است.
علائم شکل دستگاه گوارشروزها گاستروپلاز با آهسته یا، برعکس، خالی سریع معده، که می تواند مشکلات را در انتخاب درمان انسولین ایجاد کند، از آنجایی که زمان و حجم مکش کربوهیدرات ها به طور مبهم متفاوت است؛ آتون مری، ریفلاکس-اسفوژیت، دیسفاژی؛ اسهال آب برای فرم urogenitalپایین آن توسط Atony از ادرار و مثانه مشخص می شود، که منجر به گرایش به عفونت های ادراری می شود؛ اختلال نعوظ (حدود 50٪ بیماران مبتلا به SD)؛ انزال رتروگراد.
سایر تظاهرات احتمالی روز رویشی - نقض توانایی تشخیص هیپوگلیسمی، نقض عملکرد دانش آموز، نقض عملکرد غدد عرق (آنیدروز)، آمیوتروفیری دیابتی.
تشخیصی
بررسی عصبی بیماران مبتلا به SD باید سالانه انجام شود. حداقل، این امر به این معنی است که آزمایشات با هدف شناسایی نوروپاتی سنسور ديستال. برای این منظور، برآورد حساسیت ارتعاش با استفاده از Chateneton درجه بندی شده، حساسیت لمسی با مونوفیلم، و همچنین حساسیت درجه حرارت و درد استفاده می شود. بر اساس شهادت، وضعیت سیستم عصبی گیاهی مورد مطالعه قرار گرفته است: برای تشخیص نارسایی نارسایی قلب پاراسمپاتیک، تعدادی از نمونه های عملکردی مانند اندازه گیری ضربان قلب با تنفس عمیق با ارزیابی تغییرات استفاده می شود
ریتم قلب و نمونه Waltasalva؛ یک نمونه ortostatic برای تشخیص نارسایی ناشی از ناکامی سمپاتیک قلب استفاده می شود.
تشخیص های افتراقی
نوروپاتی دیگر پیدایش (الکل، اوریمی، با کم خونی 12 و غیره). تشخیص اختلال عملکرد یک یا چند عضو به عنوان یک نتیجه از نوروپاتی گیاهی تنها پس از محرومیت از آسیب شناسی ارگانیک ایجاد می شود.
رفتار
1. بهینه سازی شکر درمان.
2. مراقبت از پاها (بند 7.8.5 را ببینید).
3. اثربخشی داروهای نوروتروپیک (اسید α- لیپوئیک) در تمام مطالعات تایید نشده است.
4. علائم درمان (بیهوشی، سیلستنافیل با اختلال نعوظ، floccortisis با هیپوتانسیون اوراستاتیک، و غیره).
پیش بینی
در مراحل اولیه، روز می تواند در برابر پس زمینه جبران خسارت SD به عقب برگردد. پایین در 80٪ از بیماران مبتلا به ضایعات اولسراتیو تعیین می شود و قطع عضو اصلی خطر است
7.8.5. سندرم پا دیابتی
سندرم پا دیابتی(SDS) - حالت پاتولوژیک پا در SD ناشی از پس زمینه آسیب به اعصاب محیطی، پوست و بافت های نرم، استخوان ها و مفاصل و آشکار شدن با زخم های حاد و مزمن، ضایعات مفصلی استخوان و فرایندهای گلودرد ( جدول 7.21).
علت و پاتوژنز
پاتوژنز SDS چند بعدی است و با ترکیبی از اختلالات نوروپاتیک و پرفیوژن با تمایل به عفونت نشان داده شده است. بر اساس غلبه بر پاتوژنز یک یا چند عامل ذکر شده، 3 فرم اصلی
جدول. 7.21.سندرم پا دیابتی
 I. فرم نوروپاتیک(60-70 %):
I. فرم نوروپاتیک(60-70 %):
بدون استئوآرتروپاتی؛
با استئوآرتروپاتی دیابتی.
دوم فرم تکمیلی (مخلوط)(15-20 %).
III شکل ایسکمیک(3-7 %).
شکل نوروپاتیک SDS. در نوروپاتی دیابتی، بخش های دفاعی طولانی ترین اعصاب در درجه اول تحت تاثیر قرار می گیرند. کمبود طولانی از تحرکات طوفان منجر به هیپوتروفی پوست، استخوان ها، لیگامنت ها، تاندون ها و عضلات می شود. نتیجه هیپوتروفی ساختارهای اتصال، تغییر شکل پا با توزیع مجدد غیر فیزیولوژیکی بار مرجع و افزایش بیش از حد آن در مناطق جداگانه است. در این مکان ها، به عنوان مثال در ناحیه سرهای پروجکشن از استخوان های کراوات، ضخیم شدن پوست و تشکیل هیپرکراتوز اشاره شده است. فشار ثابت بر روی این مناطق منجر به اتولیز التهابی بافت های نرم بافتی می شود که پیش نیازهای خود را برای تشکیل یک نقص زخمی ایجاد می کند. به عنوان یک نتیجه از آتروفی و \u200b\u200bاختلال، پوست خشک می شود، آسان به ترک است. با توجه به کاهش حساسیت درد، بیمار اغلب به تغییرات رخ نمی دهد. این نمی تواند به موقع ناراحتی کفش را تشخیص دهد، که منجر به تشکیل اسکواپ ها و ذرت ها می شود، معرفی بدن های خارجی، زخم های کوچک در مکان های ترک خوردگی را متوجه نمی شود. این وضعیت اختلال حساسیت عمیق را تشدید می کند، که در نقض راه رفتن، فیلم های نامناسب ظاهر می شود. اغلب نقص زخمی با استافیلوکوک، استرپتوکوک، باکتری گروه روده آلوده است. اغلب به فلور بی هوازی می پیوندد. استئوآرتروپاتی نوروپاتیک ناشی از تغییرات شدید دیستروفیک در بونستوسرت پا (پوکی استخوان، استئولیز، هیپروستوز) است.
فرم SDS Ischemic این یک نتیجه از آترواسکلروز از شریان های اندام های پایین تر است که منجر به نقض جریان اصلی خون می شود، I.E. این یکی از انواع Macroangiopathy دیابتی است.
همهگیرشناسی
SDS در 10-25٪ مشاهده شده است، و بر اساس برخی داده ها، در یک فرم یا دیگری، در 30-80٪ از بیماران مبتلا به دیابت. در ایالات متحده، هزینه های سالانه در درمان بیماران مبتلا به SD C CDS 1 میلیارد دلار است.
تظاهرات بالینی
برای شکل نوروپاتیکSDS دو نوع مکرر ضایعات را تخصیص می دهد: زخم نوروپاتیک و استئوآرتروپاتی (با توسعه
 شکل. 7.12.زخم نوروپاتیک با سندرم پا دیابتی
شکل. 7.12.زخم نوروپاتیک با سندرم پا دیابتی
 شکل. 7.13.مشترک Warcot با سندرم پا دیابتی
شکل. 7.13.مشترک Warcot با سندرم پا دیابتی
شارکو مفصل) زخم های نوروپاتیک،به طور معمول، در منطقه ای از فواصل تک تک و فتوال قرار گرفته است، به عنوان مثال در بخش های پا که بیشترین فشار را تجربه می کنند (شکل 7.12).
تغییرات مخرب دستگاه پا پاور می تواند طی چند ماه پیشرفت کند و منجر به تغییر شکل شدید استخوان شود - arthropathy Osteo دیابتیو تشکیل مفصل Charcotدر عین حال، پا به طور رسمی با "کیسه ای با استخوان" مقایسه می شود
برای شکل ایسکمیک SDS
چرم در پاها سرد، رنگ پریده یا سیاناتیک است؛ کمتر اغلب یک سایه صورتی قرمز از گسترش مویرگ های سطحی در پاسخ به ایسکمی دارد. نقص های زخمی با توجه به نوع نكروز آکرول بوجود می آیند - بر روی نکات انگشتان دست، سطح لبه پاشنه ها (شکل 7.14).
پالس بر روی شریان های پای پا، شریان های پاپلیتیک و فمورال ضعیف است یا قابل لمس نیست.
در موارد معمول، بیماران شکایات را به "تقاطع کروم" تحمیل می کنند. شدت ضایعات ایسکمیک اندام توسط سه عامل اصلی تعیین می شود: شدت تنگی، توسعه جریان خون وثیقه، شرایط سیستم انعقاد خون.
تشخیصی
بازرسی از پا بیمار SD باید هر بار در طول بازدید از دکتر، نه کمتر از یک بار در نیم سال ساخته شود. تشخیص SDS شامل موارد زیر است:
 شکل. 7.14.نکروز عضلانی با شکل ایسکمیک سندرم پا دیابتی
شکل. 7.14.نکروز عضلانی با شکل ایسکمیک سندرم پا دیابتی
بازرسی از پاها؛
ارزیابی وضعیت عصبی - انواع مختلف حساسیت، رفلکس تاندون، الکترومیوگرافی؛
ارزیابی وضعیت جریان خون شریانی - آنژیوگرافی، داپلرمتری، داپلرگرافی؛
اشعه ایکس از مفاصل متوقف و مچ پا؛
بررسی باکتری شناسی تخلیه زخم.
تشخیص های افتراقی
این عملیات با فرآیندهای زخم در پایه های یک پیدایش دیگر، و همچنین سایر بیماری های انسداد عروق اندام های پایین تر و آسیب شناسی مفاصل پا انجام می شود. علاوه بر این، اشکال بالینی SDS باید متفاوت باشد (جدول 7.22).
رفتار
رفتار نوروپاتیک آلودهفرم های SDS شامل مجموعه ای از رویدادهای زیر است:
بهینه سازی جبران CD، به عنوان یک قاعده، افزایش دوز انسولین و با SD-2 - انتقال به آن؛
درمان آنتی بیوتیک سیستمیک؛
تخلیه کامل پا (این ممکن است باعث بهبود زخم زخم در عرض چند هفته)؛
پردازش محلی زخم با حذف بخش های hyperkeratose؛
مراقبت از پاها، انتخاب صحیح و پوشیدن کفش های ویژه. درمان محافظه کارانه به موقع انجام می شود
اجتناب از مداخله عملیاتی در 95٪ موارد.
جدول. 7.22.تشخیص دیفرانسیل فرم های SDS بالینی
 رفتار ایسکمیکفرم های SDS شامل موارد زیر است:
رفتار ایسکمیکفرم های SDS شامل موارد زیر است:
بهینه سازی جبران CD، به عنوان یک قاعده، افزایش دوز انسولین و با SD-2 - انتقال به آن؛
در غیاب ضایعات زخم نئوپروتیک، ergotherapy (پیاده روی 1-2 ساعت در روز کمک به توسعه جریان خون وثیقه)؛
عملیات Revascarcularization بر روی عروق آسیب دیده؛
درمان محافظه کارانه: ضد انعقادی، آسپرین (تا 100 میلی گرم در روز)، در صورت لزوم - فیبرینولیتیک، آماده سازی پروستاگلاندین E1 و پروستات سیکلین.
در توسعه ضایعات گسترده ای-نئوپوتیک، با تمام انواع VDS، قطع عضو مطرح شده است.
پیش بینی
از 50 تا 70 درصد از تعداد کل تحریک پاها توسط بیماران مبتلا به دیابت به حساب می آیند. قطع عضو در بیماران مبتلا به SD 20-40 برابر بیشتر از افراد بدون دیابت تولید می شود.
7.9. دیابت شکر و بارداری
دیابت بارداری(GSD) نقض تحمل گلوکز است، برای اولین بار در دوران بارداری نشان داده شده است (جدول 7.23). این تعریف حذف نمی کند که آسیب شناسی مبادله کربوهیدرات می تواند پیش از شروع حاملگی باشد. GHS باید از شرایطی که یک زن مبتلا به دیابت تشخیص داده شده (به علت سن، اغلب از SD-1)، بارداری متمایز باشد، متمایز شود.
علت و پاتوژنز
GES شبیه به کسانی است که در SD-2 هستند. سطح بالایی از استروئید های تخمدان و جفتی، و همچنین افزایش شکل گیری کورتیزول قشر آدرنال، در دوران بارداری به توسعه مقاومت به انسولین فیزیولوژیکی منجر می شود. توسعه DV ها با این واقعیت مرتبط است که مقاومت به انسولین، به طور طبیعی در دوران بارداری رشد می کند و بنابراین نیاز به افزایش انسولین در افراد مبتلا به آن بیش از توانایی عملکردی β- سلول های PJZ است. پس از تحویل با بازگشت روابط هورمونی و متابولیک به سطح اولیه، معمولا عبور می کند.
جدول. 7.23.دیابت بارداری
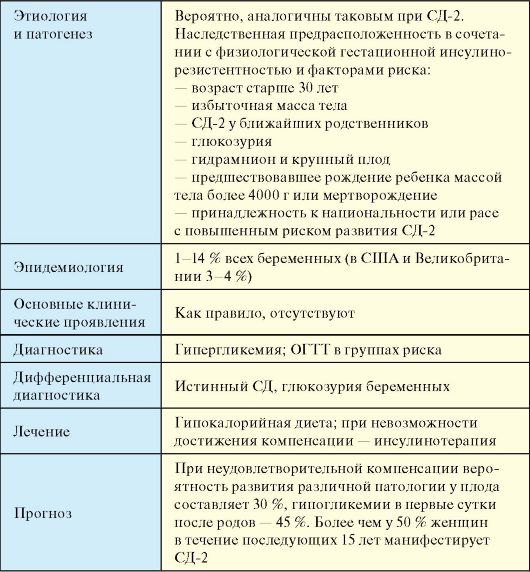 GSD معمولا در وسط 2 سه ماهه، بین 4 تا 8 ماه بارداری توسعه می یابد. اکثریت قریب به اتفاق بیماران دارای وزن بیش از حد بدن و تاریخچه AE هستند. عوامل خطر GDG، و همچنین گروه های زنان با خطر کم توسعه DV ها، در جدول نشان داده شده است. 7.24.
GSD معمولا در وسط 2 سه ماهه، بین 4 تا 8 ماه بارداری توسعه می یابد. اکثریت قریب به اتفاق بیماران دارای وزن بیش از حد بدن و تاریخچه AE هستند. عوامل خطر GDG، و همچنین گروه های زنان با خطر کم توسعه DV ها، در جدول نشان داده شده است. 7.24.
جدول. 7.24.عوامل خطر برای توسعه دیابت حاملگی
 هیپرگلیسمی مادر منجر به هیپرگلیسمی در سیستم گردش خون کودک می شود. گلوکز به راحتی به جفت نفوذ می کند و به طور مداوم به میوه خون مادر حرکت می کند. حمل و نقل فعال اسیدهای آمینه و انتقال اجسام کتون میوه نیز رخ می دهد. در مقایسه با این انسولین، گلوکاگون و اسیدهای چرب آزاد مادر در خون جنین سقوط نمی کنند. در 9-12 هفته اول بارداری، جنین PJZ هنوز انسولین خود را تولید نکرده است. این زمان مربوط به فاز ارگانوژنز جنین است، زمانی که ناهنجاری های مختلف توسعه (قلب، ستون فقرات، نخاع، گاسترویر) را می توان در هیپرگلیسمی دائمی مادر تشکیل داد. از زمان 12 هفته بارداری، جنین PJZ شروع به تولید انسولین می کند و در پاسخ به هیپرگلیسمی، هیپرتروفی واکنشی و هیپرپلازی های بتا-سلول های بتا PJZ جنین توسعه می یابد. با توجه به hyperinsulinmia، ماکروها جنین در حال توسعه است، و همچنین ظلم و ستم سنتز لسیتین، که فرکانس بالا توسعه ناراحتی تنفسی را در نوزادان توضیح می دهد. به عنوان یک نتیجه از هیپرپلازی سلول های بتا و hyperinsulinmia، تمایل به هیپوگلیسمی سنگین و طولانی به نظر می رسد.
هیپرگلیسمی مادر منجر به هیپرگلیسمی در سیستم گردش خون کودک می شود. گلوکز به راحتی به جفت نفوذ می کند و به طور مداوم به میوه خون مادر حرکت می کند. حمل و نقل فعال اسیدهای آمینه و انتقال اجسام کتون میوه نیز رخ می دهد. در مقایسه با این انسولین، گلوکاگون و اسیدهای چرب آزاد مادر در خون جنین سقوط نمی کنند. در 9-12 هفته اول بارداری، جنین PJZ هنوز انسولین خود را تولید نکرده است. این زمان مربوط به فاز ارگانوژنز جنین است، زمانی که ناهنجاری های مختلف توسعه (قلب، ستون فقرات، نخاع، گاسترویر) را می توان در هیپرگلیسمی دائمی مادر تشکیل داد. از زمان 12 هفته بارداری، جنین PJZ شروع به تولید انسولین می کند و در پاسخ به هیپرگلیسمی، هیپرتروفی واکنشی و هیپرپلازی های بتا-سلول های بتا PJZ جنین توسعه می یابد. با توجه به hyperinsulinmia، ماکروها جنین در حال توسعه است، و همچنین ظلم و ستم سنتز لسیتین، که فرکانس بالا توسعه ناراحتی تنفسی را در نوزادان توضیح می دهد. به عنوان یک نتیجه از هیپرپلازی سلول های بتا و hyperinsulinmia، تمایل به هیپوگلیسمی سنگین و طولانی به نظر می رسد.
همهگیرشناسی
CD از 0.3٪ از تمام زنان سن باروری رنج می برند، 0.2-0.3٪ از زنان باردار در حال حاضر در ابتدا بیمار هستند و در 1-14٪ از حاملگی ها، GSD در حال توسعه یا نشان دادن SD واقعی است. شیوع SDS در جمعیت های مختلف متفاوت است، بنابراین، در ایالات متحده، حدود 4 درصد از زنان باردار (135 هزار مورد در سال) تشخیص داده می شود.
تظاهرات بالینی
با GPS گم شده اند ممکن است نشانه های غیر اختصاصی از عدم توانایی SD وجود داشته باشد.
تشخیصی
تعیین سطح قند خون بر روی یک معده خالی به تمام زنان باردار در یک آزمایش خون بیوشیمیایی نشان داده شده است. زنان متعلق به گروه ریسک (جدول 7.24)، نشان می دهد تست گلوکز خوراکی(OGTT). گزینه های زیادی برای زنان باردار آن وجود دارد. ساده ترین آنها به قوانین زیر اشاره می کند:
3 روز قبل از معاینه، زن بر روی تغذیه عادی است و به فعالیت بدنی معمولی پایبند می شود؛
آزمون در صبح بر روی معده خالی انجام می شود، پس از گرسنگی شب حداقل 8 ساعت؛
پس از مصرف خون، یک زن معده خالی یک راه حل برای 5 دقیقه می نوشند، که شامل 75 گرم گلوکز خشک در 250 تا 300 میلی لیتر آب حل می شود؛ دوباره تعیین سطح گلیسمی پس از 2 ساعت انجام می شود.
تشخیص GSD به موارد زیر تنظیم شده است شاخص:
گلوکز جامد خون (وریدی، مویرگی) معده خالی\u003e 6.1 mmol / l یا
پلاسمای گلوکز خون وریدی ≥ 7 mmol / l یا
گلوکز خون مویرگی یا پلاسمای خون وریدی 2 ساعت پس از بارگیری 75 گرم گلوکز ≥ 7.8 mmol / l.
اگر یک زن متعلق به گروه ریسک باشد، نتایج این مطالعه مربوط به هنجار است، آزمون در هفته 24-28 هفته بارداری دوباره استفاده می شود.
تشخیص های افتراقی
GSD و SD واقعی؛ گلوکوزوری زنان باردار.
رفتار
خطر ابتلا به مادر و جنین، و همچنین رویکردهای درمان SD و ویژگی های کنترل آن با GDG و با SD واقعی یکسان است. عوارض دیررس SD در دوران بارداری می تواند به طور قابل توجهی پیشرفت کند، اما با جبران کیفیت بالا برای نشانه های SD برای قطع حاملگی. یک زن مبتلا به SD (به عنوان یک قاعده، ما در مورد SD-1 صحبت می کنیم)، باید بارداری در سن جوان، زمانی که خطر ابتلا به عوارض کمترین داشته باشد، باید یک بارداری را برنامه ریزی کنیم. اگر بارداری برنامه ریزی شده باشد، توصیه می شود که تماس را لغو کنید
چند ماه پس از دستیابی به جبران بهینه به دست آمد. ضد عفونی برای برنامه ریزی بارداری، نفروپاتی شدید با نارسایی کلیوی پیشرفته، سی دی های شدید، رتینوپاتی شدید پرولیفراتیو، عدم اصلاح، کتواسییدوز در اوایل بارداری (بدن کتون عوامل تراتوژنیک) است.
هدف از درمانGSD و SD واقعی در دوران بارداری، دستیابی به شاخص های آزمایشگاهی زیر است:
گلیسمی اسپول< 5-5,8 ммоль/л;
گلیسمی 1 ساعت بعد از غذا< 7,8 ммоль/л;
گلیسمی 2 ساعت پس از غذا< 6,7 ммоль/л;
مقدار متوسط \u200b\u200bمشخصات گلیسمی روزانه< 5,5 ммоль/л;
سطح HBA1C با نظارت ماهانه، مانند سالم (4-6٪).
با استفاده از SD-1، به عنوان خارج از حاملگی، یک زن باید درمان انسولین شدید را دریافت کند، اما سطح گلیسمی در دوران بارداری توصیه می شود 7-8 بار در روز ارزیابی شود. اگر غیر ممکن است برای به دست آوردن غرامت نوروگلیسمی در برابر پس زمینه تزریق عادی، لازم است که ترجمه بیمار به درمان انسولین را با استفاده از تلگراف انسولین در نظر بگیریم.
در مرحله اول درمان GSDرژیم غذایی درمانی اختصاص داده شده است، که شامل محدود کردن کالراژ روزانه حدود 25 کیلوکالری بر کیلوگرم وزن واقعی است که عمدتا به دلیل ساده ترین چربی های کربوهیدرات و حیوانی و همچنین گسترش اعمال فیزیکی است. اگر در برابر پس زمینه رژیم غذایی درمانی، امکان دستیابی به اهداف درمان وجود نداشته باشد، بیمار باید درمان شدید انسولین را تعیین کند. هر گونه داروهای شکر قرص (TSP) در دوران بارداری منع مصرفدرمان انسولین به نظر می رسد لازم است برای ترجمه حدود 15٪ از زنان.
پیش بینی
با جبران خسارت نامطلوب DV و SD در دوران بارداری، احتمال آسیب شناسی مختلف در جنین 30٪ است (خطر 12 برابر بیشتر از جمعیت کلی) است. بیش از 50٪ از زنان، که در دوران بارداری، GDS شناسایی شد، برای 15 سال آینده، SD-2 را نشان داد.
در پیشگیری از بیماری های چند فاکتوریل با یک بیماری ارثی، که EDF لینک لازم استمشاوره پزشکی و ژنتیکی. وظیفه اصلی مشاوره پزشکی و ژنتیک تعیین خطر ابتلا به بیماری و توضیح معنای آن در فرم مقرون به صرفه است. هنگامی که SD به توصیه های پزشکی و ژنتیکی، همسران اغلب معتاد هستند که به دلیل وجود این بیماری در کودکان قبلی یا در میان همسران خود و / یا بستگان آنها معتاد هستند که اغلب معتاد به ارزیابی خطر ابتلا به بیماری در کودکان آینده هستند.مطالعات جمعیتی-ژنتیک مجاز به محاسبه آن است سهم عوامل ژنتیکی در توسعه SD با60-80٪ قرار می گیرد. در این راستا، ارتباط اولیه و آینده مشاوره پزشکی و ژنتیکی خویشاوندان بیماران مبتلا به SD را به دست می آورد.
مسائل اصلی که معمولا شما باید با پزشک مواجه شوید، مربوط به خطر توسعه SD است فرزندان بیشتری دارند یا خواهران برادرانصبور، توانایی طبقه بندی آن نیز هست پیش بینی در تیتوخلاصه ای از اعضای خانواده آینده (برنامه ریزی شده).
خانواده های مشاوره ای از بیماران مبتلا به نوع نوع نوع 1 شامل چندین مرحله به طور کلی پذیرفته شده است که ویژگی های خاص خود را برای این مشروط دارند.
11.1 مراحل مشاوره
مرحله اول مشاوره - خلاصه ای از تشخیص بیماری.
معمولا تشخیص دیابت نوع 1 در سن کودکان و جوانان، مشکلات را نشان نمی دهد. با این حال، در حضور SD، سایر اعضای خانواده نیاز به تایید نوع دیابت دارند که در بعضی موارد ممکن است یک کار دشوار باشد و به پزشک نیاز داشته باشد تا به دقت جمع آوری آنامنز یک بیمار را به دقت جمع آوری کند. تشخیص دیفرانسیل بین دو نوع اصلی SD (1 و 2) بر اساس معیارهای پذیرفته شده به طور کلی انجام می شود.
اثبات شده با مطالعات ژنتیکی، ناهمگونی ژنتیکی، ناهمگونی ژنتیکی دو نوع اصلی SD شهادت می دهد به استقلال بیولوژیکی و استقلال ارث. این به این معنی است که کسانی که در بیماران مبتلا به سجده ای با انواع سی دی 2 موجود هستند، به صورت تصادفی هستند و نباید در هنگام ارزیابی ریسک خانوادگی مورد توجه قرار گیرند.
هنگام انجام مشاوره پزشکی و ژنتیکی، ضروری است که سندرم های ژنتیکی را که شامل دیابت می شوند، حذف کنیم، زیرا آنها توسط ارثی مونوژنیک مشخص می شوند.
مرحله دوم مشاوره - تعیین خطر توسعه بیماری در مورد اعضای خانواده موجود و فرزندان برنامه ریزی شده.
روشهای تجربی با میانگین ارزیابی ریسک دیابت به دست آمد برای اعضای خانواده های خانوادگی با بستگان با نوع 1 SD. حداکثر خطر دارای بستگان درجه خویشاوندان من (کودکان، والدین، برادران خواهر) - به طور متوسط \u200b\u200bاز 2.5-3٪ تا 5-6٪ است. ثابت شده است که فراوانی بیماری دیابت کودکان از پدران با SD 1 نوع 1 -2٪ بالاتراز مادران با نوع نوع 1.
در هر خانواده خاص، خطر ابتلا به بیماری بستگی به عوامل بسیاری دارد: تعداد بیماران و بستگان سالم، سن تظاهرات دیابت در اعضای خانواده، سن مشورت، و غیره
جدول 8
خطر تجربی برای بستگان بیماران SD نوع 1
در یک تکنیک خاص محاسبه می شود توسعه جداول ریسکSD 1. نوع بسته به تعداد بیماران و بستگان سالم و سن مشورت کنید برای خانواده های مختلف. خانواده ها، وضعیت والدین و تعداد بیماران مبتلا به SIBS در جدول 9 ارائه شده است.
سندرم داون یک بیماری نیست، این آسیب شناسی غیر ممکن است برای جلوگیری و درمان. جنین با سندرم داون در 21st جفت کروموزوم، یک کروموزوم اضافی وجود دارد، به عنوان یک نتیجه از مقدار آنها 46 نیست، اما 47. سندرم داون در یکی از نوزادان 600-1000 نوزاد پس از 35 سال دیده می شود. دلیل این که این اتفاق می افتد، کاملا روشن نیست. دکتر انگلستان جان لانگدون برای اولین بار این سندرم را در سال 1866 توصیف کرد و در سال 1959 پروفسور فرانسوی Lezhen ثابت کرد که این به دلیل تغییرات ژنتیکی است.
شناخته شده است که کودکان نیمی از کروموزوم از مادر و نیمی از پدر دریافت می کنند. از آنجاییکه یک روش موثر برای درمان سندرم وجود ندارد، این بیماری غیر قابل درمان است، شما می توانید اقدامات را انجام دهید و اگر مایل به تولد فرزند سالم هستید، با مشاوره پزشکی و ژنتیکی تماس بگیرید، جایی که بر اساس کروموزومی تجزیه و تحلیل، والدین تعیین می شود، کودک سالم یا با سندرم داون متولد خواهد شد.
به تازگی، چنین کودکان اغلب متولد می شوند، آنها آن را با ازدواج دیر، با برنامه ریزی بارداری 40 ساله مرتبط می کنند. همچنین اعتقاد بر این است که اگر مادربزرگ پس از 35 سالگی فرزند خود را به دنیا آورد، نوه ها می توانند با سندرم داون متولد شوند. اگر چه تشخیص قبل از زایمان یک فرآیند بررسی پیچیده است، اجرای آن بسیار ضروری است تا بتواند بارداری را متوقف کند.
سندرم داون چیست؟ این معمولا می تواند با تاخیر توسعه موتور همراه باشد. چنین کودکان دارای نقایص قلب مادرزادی، آسیب شناسی توسعه دستگاه گوارش هستند. 8٪ از بیماران مبتلا به سندرم داون بیمار مبتلا به لوسمی هستند. درمان پزشکی می تواند فعالیت ذهنی را تحریک کند، عدم تعادل هورمونی را نرمال کند. با کمک روش های فیزیوتراپی، ماساژ، ژیمناستیک درمانی، می توانید به کودک کمک کنید تا مهارت های لازم برای خدمات خود را بدست آورید. سندرم داون با نقض ژنتیکی همراه است، اما همیشه منجر به نقض رشد جسمی و ذهنی کودک نمی شود. چنین کودکان، و در آینده بزرگسالان می توانند در تمام حوزه های زندگی شرکت کنند، بعضی از آنها تبدیل به بازیگران، ورزشکاران می شوند و می توانند در امور عمومی شرکت کنند. چگونه یک فرد با این تشخیص توسعه خواهد یافت بستگی زیادی به محیط زیست دارد که در آن رشد می کند. شرایط خوب، عشق و مراقبت به توسعه کامل کمک می کند.
جدول خطر سندرم داوون، با سن
احتمال سندرم داون به سن مادر بستگی دارد، اما می توان آن را با آزمون ژنتیکی در مراحل اولیه حاملگی نشان داد، و در بعضی موارد اولتراسوند. احتمال ابتلا به سندرم داون کودک در هنگام تولد کمتر از مراحل اولیه بارداری است، زیرا بعضی از میوه ها با سندرم داون زنده ماندند.

چه ریسک کم است، و چه چیزی بالا است؟
در اسرائیل، خطر ابتلا به سندرم داون بالا است، اگر بالاتر از 1: 380 باشد (0.26٪). هر کسی که در این گروه خطر قرار دارد، باید توسط آب های آستانه مورد بررسی قرار گیرد. این خطر برابر با خطر برای کسانی است که باردار در سن 35 سالگی و بالاتر است.
خطر کمتر از 1: 380 کم است.
اما باید در ذهن داشته باشید که این مرزها می توانند شناور باشند! به عنوان مثال، در انگلستان، سطح بالایی از خطر بالاتر از 1: 200 (0.5٪) است. این به خاطر این واقعیت است که برخی از زنان خطر ابتلا به 1 تا 1000 نفر را در معرض خطر قرار می دهند، و دیگر 1 تا 100 - کم، از آنجا که با چنین خطر هایی برای تولد یک کودک سالم برابر با 99٪ است.
عوامل خطرساز سندرم، ادواردز، Patau
عوامل خطر اصلی سن (به ویژه به طور قابل توجهی برای سندرم داون)، و همچنین تاثیر تابش، برخی از فلزات سنگین است. باید در نظر داشته باشید که حتی بدون عوامل خطر، میوه ممکن است پاتولوژی داشته باشد.
همانطور که از گراف دیده می شود، وابستگی خطر ابتلا به ریسک از سن، مهمترین سندرم داون است و کمتر برای دو تریسومی دیگر قابل توجه است:

غربالگری خطر سندرم داون
تا به امروز، همه زنان باردار علاوه بر تکیه بر تجزیه و تحلیل، توصیه می شود تا آزمایش غربالگری را برای شناسایی میزان خطر ابتلا به سندرم داون با تولد یک کودک و زخم های جنینی مادرزادی انجام دهید. نظرسنجی مولد در 11 هفته + 1 روز یا در هفته 13 + 6 روز با یک کواکسیکو-تاریکی جنین از 45 میلیمتر تا 84 میلی متر رخ می دهد. یک زن باردار می تواند تحت بررسی قرار گیرد و از اولتراسوند خاص برای این استفاده کند.
تشخیص دقیق تر با استفاده از بیوپسی کوریون وین و مطالعه مایع آمنیوتیک، که با استفاده از یک سوزن مخصوص به طور مستقیم از حباب میوه بسته شده است، ساخته شده است. اما هر زن باید بداند که چنین روش هایی با خطر ابتلا به عوارض حاملگی مانند سقط جنین، عفونت جنین، توسعه شنوایی شنوایی در کودک و خیلی بیشتر مرتبط هستند.
غربالگری کامل ترکیبی از سه ماهه I - II بارداری به شما اجازه می دهد تا بدبختی های مادرزادی را در جنین شناسایی کنید. چه چیزی شامل این آزمون است؟ اول، یک مطالعه سونوگرافی در 10-13 هفته بارداری مورد نیاز است. محاسبه خطر این است که تعیین حضور استخوان بینی، عرض دهانه رحم جنین، جایی که مایع زیر جلدی در اولین دوره بارداری تجمع می یابد.
در عرض چند ثانیه، آزمایش خون بر روی گونادوتروپین کوریونیک در 10-13 هفته و در فوتئوپروتئین آلفا در 16-18 هفته گرفته می شود. داده های غربالگری ترکیبی توسط یک برنامه کامپیوتری خاص پردازش می شود. دانشمندان یک روش غربالگری جدید را پیشنهاد دادند - ترکیب ارزیابی نتایج به دست آمده در طی مطالعات در اولین و در سه ماهه دوم. این به ما اجازه می دهد تا یک ارزیابی ریسک یک خطر ابتلا به سندرم داون را در دوران بارداری تضمین کنیم.
برای سه ماهه اول، نتایج تعیین RARR-A و اندازه گیری ضخامت فضای یقه استفاده می شود، و برای سه ماهه دوم - ترکیبی از AFP، استریول غیر مرتبط، XG و مهار کننده استفاده می شود. استفاده از یک ارزیابی انتگرال برای بررسی غربالگری اجازه می دهد پس از مداخلات تهاجمی برای کاهش فرکانس قطع بارداری برای میوه ها با کاروتیپ طبیعی با توجه به نتایج تشخیص سیتوژنتیک.
تست انتگرال و بیوشیمیایی برای سندرم غربالگری غربالگری به شما امکان می دهد تا موارد بیشتری از ناهنجاری های کروموزومی را شناسایی کنید. این امر موجب جلوگیری از وقفه های حاملگی نامطلوب ناشی از آمنیوسنتس یا بیوپسی کوروین می شود.
ویرایشگر کارشناس: Mochalov Pavel Alexandrovich | د درمانگر
تحصیلات: موسسه پزشکی مسکو. I. M. Sechenov، تخصص - "مورد درمانی" در سال 1991، در سال 1993 "بیماری های حرفه ای"، در سال 1996 "درمان".
ژنتیک دیابت
پیش بینی نوع 1 SD 1 در گروه های پر خطر
T.V.Nikonova، I.I. پدربزرگ، JI.P. Alekseev، M.N. boldyreva، o.m. Smirnova، I.V. Dubinkin *.
مرکز علمی غدد درون ریز I (گوزن - Acads. Ramn I. I. I. Devov) Ramna، I * SSC "موسسه ایمونولوژی" I (گوزن - Acad. Ramna P.m. Khaitov) M3 فدراسیون روسیه، مسکو. من
در حال حاضر، افزایش میزان بروز SD 1 در سراسر جهان وجود دارد. این به دلیل تعدادی از عوامل، از جمله افزایش امید به زندگی بیماران مبتلا به دیابت به دلیل بهبود کمک های تشخیصی و درمانی، افزایش باروری و بدتر شدن شرایط محیطی است. کاهش میزان بروز SD را می توان با اقدامات پیشگیرانه، پیش بینی و جلوگیری از توسعه بیماری انجام داد.
مستعد ابتلا به نوع SD 1 ژنتیکی تعیین می شود. بروز نوع 1 نوع 1 توسط تعدادی از ژن ها کنترل می شود: ژن انسولین بر روی کروموزوم 11P15.5 (YUOM2)، ژن های کروموزوم \\ \\ C (YUOM4)، 6T (YUOM5). بزرگترین اهمیت از نشانگرهای شناخته شده ژنتیک نوع 1 نوع 1 ژنهای منطقه NAA بر روی کروموزوم 6P 21.3 (SEW1)؛ تا 40٪ از استعمار ژنتیکی به نوع SD 1 با آنها همراه است. هیچ منطقه ژنتیکی دیگر خطر ابتلا به بیماری را که قابل مقایسه با Na است، تعیین نمی کند.
ریسک بالا نوع توسعه نوع نوع 1 توسط انواع آلل ژن های هسته ای تعیین می شود: دسامبر 1 * 03، * 04؛ OOA1 * 0501، * 0301، OOB1 * 0201، * 0302. 95٪ از بیماران با 1 نوع SD دارای آنتی ژنهای AMA * 3 یا 011 * 4 هستند و از 55 تا 60 درصد هر دو آنتی ژن دارند. Allel OOB1 * 0602 به ندرت در یک نوع نوع 1 مواجه می شود و به عنوان اعتراض محسوب می شود.
تظاهرات بالینی دیابت پیش از یک دوره پنهان، مشخص شده با حضور نشانگرهای ایمنی سلولی جزیره؛ این نشانگرها با تخریب پیشرونده همراه است.
بنابراین، برای اعضای خانواده با موارد پیش از بیماری بیماری SD 1، پیش بینی بیماری به ویژه مهم است.
هدف از این کار، تشکیل گروه های توسعه پر خطر بالا در جمعیت روسیه ساکنان مسکو بر اساس مطالعه نشانگرهای ژنتیکی، ایمونولوژیک و متابولیک دیابت با استفاده از رویکرد خانوادگی بود.
مواد و روش های تحقیق
26 خانواده مورد بررسی قرار گرفت که در آن یکی از والدین بیمار 1 نوع SD 1، که 5 خانواده "هسته ای" هستند (تنها 101 نفر). تعداد اعضای خانواده مورد بررسی از 3 تا 10 نفر بود. بیماران SD 1 نوع پدران - 13، بیماران مبتلا به MAT 1 نوع مادران نیز 13 ساله هستند. خانواده هایی که هر دو والدین بیمار خواهند شد 1 نوع، هیچ.
37 نسل از بیماران مبتلا به SD 1 بدون تظاهرات بالینی بیماری مورد بررسی قرار گرفتند که از آن 16 -Gen-genus 21 مرد هستند. سن از فرزندان مورد بررسی از 5 تا 30 سال متغیر بود. توزیع نسل های مورد بررسی توسط سن در جدول ارائه شده است. یکی
میز 1
سن کودکان مورد بررسی (فرزندان)
سن (سال ها) تعداد
در خانواده های مبتلا به مادران SD، 17 کودک مورد بررسی قرار گرفتند (8 دختر، 9 پسر)، در خانواده های بیماران مبتلا به دیابت - 20 کودک (8 دختر، 12 پسر).
یک عامل آنتیل (3 سلول (ICA) به دو روش تعیین شد: 1) بر روی کریستال پانکراس مرد I (0) گروه های خون در واکنش ایمونوفلورسانس غیر مستقیم؛ 2) در Islettest "Islettest" شرکت "Biomerica". یک آنتی بادی برای انسولین (IAA) در آزمایش آنزیمی "Eslettest" Biomerica تعیین شد. تعریف آنتی بادی های DGC با استفاده از مجموعه های استاندارد "دیپشت های ضد گاد" شرکت "Boehringer Mannheim" انجام شد.
تعریف C-peptide با استفاده از مجموعه های استاندارد شرکت "Sorrin" (فرانسه) انجام شد.
تایپ HLA از بیماران مبتلا به SD و خانواده های آنها بر روی سه ژن انجام شد: DRB1، DQA1 و DQB1 با استفاده از پرایمرهای Cyphic - "" با استفاده از یک واکنش زنجیره ای پلیمریزاسیون (PCR).
انتشار DNA از لنفوسیت های خون محیطی بر اساس روش R. higuchi n. erlich (1989) با برخی از اصلاحات انجام شد: 0.5 میلی لیتر خون گرفته شده با EDTA، مخلوط شده در 1.5 میلی لیتر از لوله های میکروکنترل مانند Eppendorf با 0.5 میلی لیتر لیزر یک راه حل متشکل از 0.32 متر ساکارز، 10 میلیمتر Tris - NS1 pH 7.5، 5mm mgc12، 1٪ تریتون X-100، سانتریفیوژ شده به مدت 1 دقیقه در 10،000 دور در دقیقه، سوپرناتانت برداشته شد، و رسوبات هسته های سلول 2 بار شسته شده با بافر مشخص شده پروتئولیز بعدی در 50 میکرولیتر از یک محلول بافر حاوی 50 میلی مولار KCI، 10 میلی متر Tris-NS1 pH 8.3، 2.5 میلی متری MgCi2، 0.45٪ NP-40، 0.45٪ دوقلوهای 20 و 250 میکروگرم در میلی لیتر پروتئیناز K در 37 بود "C به مدت 20 دقیقه. پروتئیناز غیر فعال شده به حرارت در یک ترموستات حالت جامد در 95 "C به مدت 5 دقیقه. نمونه های DNA به دست آمده بلافاصله برای تایپ کردن یا ذخیره شده در -20 استفاده می شود. غلظت DNA تعریف شده توسط
فلورسانس با Hoechst 33258 در فلوریتر DNA (Hoefer، USA)، به طور متوسط \u200b\u200b50-100 میکروگرم در میلی لیتر بود. کل زمان روش جداسازی DNA 30-40 دقیقه بود.
PCR در 10 میکرولیتر مخلوط واکنش حاوی 1 میکرولیتر نمونه DNA و غلظت های زیر از اجزای باقی مانده انجام شد: 0.2 میلی متر هر DNTF (DATF، DTTF، DTTF و DGTF)، 67mm tris-hcl pH \u003d 8.8، 2.5 MM MGC12، 50 میلی مولار NaCl، 0.1 mg / ml ژلاتین، 1 میلیمتر 2-Mercaptoethanol، و همچنین 1 واحد ترمومتری DNA پلیمراز. برای جلوگیری از تغییرات غلظت مولفه های مخلوط واکنش به علت تشکیل تراکم، مخلوط واکنش با 20 میلی لیتر روغن معدنی (سیگما، ایالات متحده آمریکا) پوشیده شده بود.
تقویت در Cycler حرارتی MS2 MS2 انجام شد (JSC تکنولوژی DNA، مسکو).
تایپ کردن Locus DRB1 در 2 مرحله انجام شد. در طول دور اول، DNA ژنومی در دو لوله مختلف تقویت شد. در لوله اول لوله، یک جفت پرایمر، تمام آلل های شناخته شده ژن DRB1 را تقویت می کند، در دوازدهمین پرایمر، تکثیر تنها آلل های موجود در DR3، DR5، DR6، DR8 استفاده می شود. در هر دو مورد، دمای تقویت (برای ترموسیکلیک "MS2" با مقررات فعال) به شرح زیر بود: 1) 94 درجه سانتیگراد - 1 دقیقه؛ 2) 94 ° C - 20 C (7 دوره)، 67 "C - 2 C؛ 92 "C - 1 C (28 سیکل)؛ 65 ° C - 2 S
محصولات به دست آمده 10 بار تقسیم شده و در دور دوم در حالت دمای بعدی استفاده می شود: 92 "C - 1 C (15 دوره)؛ 64 درجه سانتیگراد - 1 ثانیه.
تایپ DQA1 Locus در 2 مرحله انجام شد. در مرحله اول، یک جفت پرایمر، تمام ویژگی های DQA1 Locus را تقویت می کند، در جفت دوم، در دومین دوره پرایمر، ویژگی های دامنه فیتکشن * 0101، * 0102، * 0103، * 0201، * 0301، * 0401، * استفاده شد 0501، * 0601.
مرحله اول با توجه به برنامه انجام شد: 94 "C - 1 دقیقه؛ 94 ° C - 20 C (7 cycles)، 58 "C - 5 C؛ 92" C - 1 C، 5 C (28 CYCLES)، 56 "C - 2 S.
محصولات تقویتی مرحله اول 10 بار تقسیم شده و در مرحله دوم استفاده می شود: 93 "C - 1 C (12 سیکل)، 62" C - 2 S.
تایپ کردن Locus DQB1 نیز در 2 مرحله انجام شد؛ یک جفت آغازگر، تمام ویژگی های Locus DQB1 را تقویت می کند، رژیم دما به شرح زیر است: 94 "C - 1 دقیقه. 94 درجه سانتیگراد - 20 ثانیه (7 سیکل)؛ 67" C - 5 S.؛ 93 ° C - 1 C (28 سیکل)؛ 65LS - 2 p.
مرحله دوم از جفت آغازگرها استفاده می شود، ویژگی تقویت کننده: * 0201، * 0301، * 0302، * 0303، * 0304، * 0305، * 04، * 0501، * 0502، * 0503، * 0601، * 0602، * 0601، * 0602/08؛ محصولات مرحله اول 10 بار تقسیم شدند و در حالت تکثیر شدند: 93 "C - 1 S. (12 سیکل)؛ 67" C - 2 S.
شناسایی محصولات تقویتی و توزیع طول آنها در نور ماوراء بنفش (310 نانومتر) پس از الکتروفورز به مدت 15 دقیقه یا 10٪ Paag، 29: 1 در یک ولتاژ 500V یا در ژل 3٪ آگارز در یک ولتاژ 300 ولت (در هر دو مورد، مسافت پیموده شده 3-4 سانتی متر بود) و رنگ آمیزی با برومید اتیدیم. به عنوان نشانگر طول، PUC19 پلاسمید PUC19 محدود کننده MSP مورد استفاده قرار گرفت.
نتایج و بحث آن
ثابت شده است که در 26 خانواده از 26 بیمار از SD 1 نوع والدین 23 نفر (88.5٪) حامل های مرتبط با DRB1 * 05-DQA1 * 0501 - DQB1 * 0201 مرتبط با DQB1 * 0201 - DQB1 * 0201؛ DRB1 * 04-DQAL * 0301-DQB 1 * 0302 یا ترکیب آنها (جدول 2). در 2 بیمار در ژنوتیپ، Allele DQB 1 * 0201 وجود دارد، همراه با یک نوع نوع 1؛ تنها 1 بیمار از این گروه ژنوتیپ DRB1 * 01/01 را کشف کرد که
توزیع ژنوتیپ ها در میان بیماران مبتلا به SD 1 والدین
01 در 1 4/4 2 E1؟ در 1 - - - -
مجموع 23 (88.5٪) مجموع 3
0і؟ B1-ROA-RO GaPlotypes شناسایی شده توسط افراد مورد نظر
oogvі ooі r)
rY با مطالعات جمعیتی با نوع نوع 1 همراه نبود، ما زیرمجموعه های B1 * 04 را تخصیص نکردیم، اگر چه پلی مورفیسم این مکان ممکن است بر خطر نوع 1 SD تاثیر بگذارد.
در ژنوتایپ نسل مستقیم بیماران مبتلا به نوع 1، مشخص شد که از 37 نفر 30 (81٪) به ارث برده شده با SD 1 از نوع ژنوتیپ های OV1 * 03، 011V1 * 04 و ترکیب آنها، در 3 نفر در ژنوتیپ همراه با SD 1 نوع آلل 1: 1 - OOA 1 * 0501، در 2 بیمار -Oov 1 * 0201. در مجموع 4 مورد از 37 مورد نظر، ژنوتیپ خنثی با توجه به نوع نوع 1 هستند.
توزیع ژنوتیپ های فرزندان در جدول نشان داده شده است. 3. در تعدادی از "آثار، اشاره شده است که بیماران پدران SD 1 اغلب باعث انتقال ژنتیک می شوند
نادرست به دیابت (به ویژه، انواع Na-01 * 4 ژنتیک) به فرزندان خود نسبت به مادر. با این حال، مطالعه در انگلستان تأثیرات اساسی جنسیت والدین را در مورد پیشگیری از هسته در کودکان تأیید نکرد. در کار ما، ما همچنین نمی توانیم چنین منظم انتقال از انتقال ژنتیکی را ذکر کنیم: از بیماران مبتلا به مادران مرتبط با نوع SD 1 ژنوتایپ NY به ارث برده 94٪ کودکان و بیماران مبتلا به پدران - 85٪.
SD، همانطور که شناخته شده است، یک بیماری چند فاحشه چند نفره است. به عنوان عوامل محیطی خارجی که نقش یک ماشه را بازی می کند، مواد غذایی در نظر گرفته می شود - مصرف پستان و دوران کودکی پروتئین شیر گاو. د
جدول 3
توزیع ژنوتیپ ها در میان کودکان که والدین آنها بیمار هستند 1 نوع
GOTYPES مرتبط با SD 1 نوع تعداد حامل های رسانه ای، با تعداد SD 1 تعداد رسانه ها مرتبط نیست
0! * در 1 4/4 4 01 * در 1 1/15 1
مجموع 30 (81٪) تنها 7 (19٪)
با دیابت تازه تشخیص داده شده، سطح بالایی از آنتی بادی ها به پروتئین شیر شیر گاو، P-lactoglobulin و آلبومین بولشیت نسبت به SIBS سالم وجود دارد که به عنوان یک عامل خطر مستقل SD در نظر گرفته می شود.
در گروهی از کودکان مورد بررسی از 37 نفر، تنها 4 نفر در تغذیه با شیر مادر تا 1 سال، 26 نفر شیر مادر را به 1.5-3 ماه، 4 تا 6 ماه، 3 در ترکیب لبنیات از هفته اول زندگی دریافت کردند. از 5 کودک مبتلا به آنتی بادی مثبت به سلول های P- سلول های P شیر مادر تا 6 ماه، 3 - تا 1.5 تا 3 ماه تغذیه شدند. سپس مخلوط کافر و لبنیات به دست آمد. بنابراین، 89٪ از کودکان مورد بررسی پروتئین شیر گاو در عصر قلب و دوران کودکی دریافت کردند که ممکن است به عنوان یک عامل خطر توسعه SD در افراد مبتلا به ژنتیک پیش بینی شود.
در خانواده های مورد بررسی، نسل های بالینی سالم با استفاده از آنتی بادی های سیتوپلاسمی، آنتی بادی ها به انسولین و DGC تعیین شدند. از 37، 5 کودک مورد بررسی در حضور آنتی بادی ها به سلول های P مثبت بودند، در حالی که همه 5 حامل حساسیت ژنتیکی به SD (جدول 4) بودند. در 3 نفر از آنها (8٪)، آنتی بادی های DGC در 1 - به Azok، در 1 - آنتی بادی به Azok یافت شد
جدول 4
Gotypes از کودکان مثبت در آنتی بادی های K (3 سلول
تعداد آنتی بادی های مثبت
و انسولین بنابراین، آنتی بادی های Azok دارای 5.4٪ از کودکان، 2 کودک مبتلا به آنتی بادی مثبت DGC، فرزندان خانواده های هسته ای هستند. سن کودکان در زمان تشخیص آنتی بادی ها در جدول نشان داده شده است. 5. برای پیش بینی SD، سطوح تیتر Azok از اهمیت زیادی برخوردار است: بالاتر از عنوان آنتی بادی، احتمال ابتلا به توسعه SD، همان چیزی است که به آنتی بادی ها به انسولین اعمال می شود. با توجه به ادبیات، سطح بالایی از آنتی بادی های DGC با سرعت پایین تر از توسعه SD (10٪ در 4 سال) نسبت به سطح پایین (50٪ در 4 سال) همراه است، شاید به دلیل سطح بالا "آنتی بادی ها به DGC نشان دهنده فعال سازی مصونیت هومورال و به میزان کمتر برای فعال سازی سلولی
جدول 5
سن کودکان مورد بررسی در زمان تشخیص آنتی بادی ها
سن کودکان مورد بررسی (سال) تعداد کودکان مثبت در آنتی بادی ها
ایمنی (نوع 1 نوع SD 1 به طور عمده به دلیل تخریب سلول های P سلول توسط لنفوسیت های سیتوتوکسی T-lymphocytes) است. ترکیبی از آنتی بادی های مختلف، سطح مطلوب پیش بینی را تضمین می کند.
در کودکان مبتلا به وزن کم بدن در هنگام تولد (کمتر از 2.5 کیلوگرم)، دیابت به طور قابل توجهی زودتر از کودکان متولد شده با جرم طبیعی رشد می کند. از داده های انامنز، این واقعیت که از 5 کودک مبتلا به آنتی بادی مثبت 2 با جرم بدن بیش از 4 کیلوگرم، 2 درصد 2.9 کیلوگرم متولد شد.
در نسل مستقیم بیماران مبتلا به نوع بیماران نوع 1، سطح پایه پپتید C تعیین شده است، تمام این شاخص ها در محدوده طبیعی (از جمله در کودکان با آنتی بادی های مثبت به سلول های R- سلول های R)، مطالعه سطح بود از پپتید C تحریک شده انجام نشد.
1. بیماران نوع نوع 1 در 88.5٪ موارد، حامل ژنوتایپ های مشتقات، OOA1 * 0501، VOV1 * 0201، OV1 * 04، BOA1 * 0301، EOB1 * 0302 یا ترکیب آنها هستند.
2. در کودکان از خانواده ها، جایی که یکی از والدین بیمار 1 نوع نمونه، در 89٪ موارد، یک نوع ژنتیکی به SD (در حضور یک والدین بیمار)، و 81٪ به طور کامل با نوع SD 1 به ارث برده می شود ژنوتیپ ها، که به آنها اجازه می دهد آنها را یک گروه از خطر بسیار بالایی از دیابت را در نظر بگیرند.
3. در میان فرزندان مستقیم بیماران مبتلا به SD 1، داشتن یک نوع ژنتیکی، آنتی بادی های مثبت به DGC در 8٪ موارد، ACOC - در 5/5٪ موارد نشان داده می شود. این کودکان نیاز به یک مطالعه تشخیصی از تیترهای آنتی بادی، گلیکموگلوبین و مطالعه ترشح انسولین دارند.
* 1 ITERATE
1. Atkinson M.A.، McLaren N.K. // n.engl.j.med.-l 994 -331. P.4281436.
2. Aanstoot H.J.، Sigurdsson E.، Jaffe M. et al // دیابتیولوژی-1994-37.
3. Baekkeskov S.، Aanstoot H.j.، Christgan S. et al // Nature-1 990-377.
4. Bain S.c.، Rowe B.R.، Barnett A.H.، Toddj.a. // دیابت-1994-43 (12). ص 1432-1468.
5. B / NG / EY P.j.، Christie M.r.، Bonifacio E.، Bonfanti R.، Shattock MW Fonte M.T.، Bottazzo C.F. // دیابت -1 994-43. ص. 1304-1310
6. Boehn B.O.، Manifras B.، Seiblerj. و همکاران دیابت- 1991-40. p.1435-1439.
7. Chern M.m.، Anderson V.E.، Barbosa J. // دیابت-1982-31. P.L 1 151118.
8. Davies J.L.، Kawaguchi Y.، بنت S.T. و همکاران // طبیعت 1994-371.
9. Erlich H.A.، Rotter J.i.، Chang J. و همکاران. // طبیعت Gen.-1993-3. p.358-364.
10. Hahl J.، Simell T.، Ilonen J.، Knip M.، Simmel O. // دیابتیولوژی -1 99841. P.79-85.
11. هریسون L.C.، Honeyman M.C.، Deazpurua H.J. و همکاران // lancet-199341. P.L 365-1369.
12. Hashimoto L.، Habita C.، Beresse J.P. و همکاران // طبیعت 1994-371. p.161-164.
1 3. Karjalainen J.، Martin J.m.، Knip M. et al // n.engl.j.med.-L 992-327. p.302-303.
14. خان N.، CoPert.T.، // دیابت مراقبت-1994-17. ص. 653-656.
15. Landin-Oisson M. Palmer J.P.، Lernmark A. et al // دیابتیولوژی-1992-40. p.l068-1 073.
16. Leslie R.D.C.، Atkinson M.A.، Notkins A.L. // دیابتیولوژی-1999-42. ص .3-14.
17. Levy-Marchal C.، Dubois F.، Neel M.، Tichetj.، Czernichow P. // دیابت-1995-44. ص .1029-1032.
1 8. Lorenzen T.، Pociot F.، Hougaard P.، Nerup J. // دیابتیولوژی-1994-37. ص .321-321.
19. Lorenzen T "Pociot F.، Stilgren L. et al // دیابتیولوژی-1998-41. ص .666-673
20. Nepom G.، Erlich H.A. // ann.Rev.Immunol. 1 991-9. ص .493-525
21. Nerup J.، Mandrup-Poulsen T.، Molvig J. // دیابت متاب. Rev.-1987-3. p.779-802.
22. Owerbach D.، Gabbay K.H. // دیابت-1995-44.P.L 32-136.
23. Pociotf. U dan.med.Bull.-L996-43. p.216-248.
24. Rewers M.، Bugawan T.L.، Norris J.M.، بلر A. و همکاران. // دیابتیولوژی-1996-39. P.807-812.
25. Rei / Onen H.، Ilonen J.، Knip N.، Akerblom H. // دیابت-1 991-40.
26. Saukkonen T.، Virtanen S.M.، Karppinen M. et al // دیابتیولوژی-1998-41. p.72-78.
27. Schatz D.، Krischerj.، Horne G. et al // J. Clin. سرمایه گذاری - 1994-93. p.2403-2407.
28. Spielman R.S.، Baker L، Zmijewski C.M. // ann. هوم ژنتیک 1980-44. ص. 135-150.
29. Thivolet C.، Beaufrere B.، Gebuhrer Y.، Catelain P.، Orgiazzi J. // دیابتیولوژی-1991 -34. P.L86-191.
30. Tillil H.، Kobberling J. :// دیابت-1982-36. p.93-99.
31. Toddj.a. تو پروس ناتل ACAD. SCI USA-1990-377. P.8560-8565.
32. Toddj.a.، Farral M. // hum.mol.gen.-5. p.1443-1448.
33. Tuomilehto J.، Zimmet P.، Mackay I.R. و همکاران // lancet-1994-343.
34. Van der Anvera B.، Van Waeyenegenge C.، Schuit F. et al. // دیابت-1995-44. p.527-530.
35. واکر A. Gudworth AG. // دیابت-1980-29. ص .1036-1039.
36. Warram J.، Krolewski A.S.، Gottlieb M.، Kahn C.R. // n.engl.j.med.-1984-311. p.149-151.
37. Ziegler A.G.، Herskowitz R.D.، جکسون R.A. ویتامین // دیابت مراقبت-1990-13. p.762-775.



